Human angiotensin-converting enzyme 2, hACE2, sits at the intersection of several medically important pathways. As a monocarboxypeptidase anchored to cell surfaces in the lung, kidney, and gut, it regulates blood pressure by converting the vasoconstrictor angiotensin II into the vasodilator angiotensin 1–7, processes bioactive peptides including apelin and des-Arg9-bradykinin, and serves as the entry receptor exploited by SARS-CoV-2. Altered hACE2 expression has been linked to cardiovascular disease, diabetes, obesity, and inflammatory bowel disease, making selective inhibition an attractive therapeutic strategy. Phage and mRNA display have already delivered potent cyclic peptide inhibitors of hACE2, including the bicyclic BCY15291 with a Ki of 0.9 nM and mRNA-derived peptide20 with a Ki of 370 nM, yet both platforms rely on iterative, difficult-to-monitor selection procedures that require downstream chemical synthesis before a binding affinity can be measured.
Researchers in the Angelini Lab at Ca' Foscari University of Venice, published in J. Med. Chem., deployed a yeast display platform they previously validated on five non-therapeutic targets to attack this clinically relevant enzyme. Five naïve libraries encoding disulfide-cyclized macrocyclic peptides, MPs, in both "one-ring" CXmC topologies (m = 7 or 9) and "two-ring" CXmCXnC topologies (m + n = 12) were pooled and screened against biotinylated recombinant hACE2. Two rounds of magnetic bead separation, preceded by negative selection against streptavidin-coated beads, enriched binders before four rounds of fluorescence-activated cell sorting refined the population further. Critically, the platform allows binding affinities to be read directly from yeast cell surfaces without synthesizing the peptides first, and the authors alternated neutravidin and streptavidin detection reagents across FACS rounds to prevent enrichment of reagent-binding artifacts. Sanger and next-generation sequencing after the fourth sort identified three consensus families.
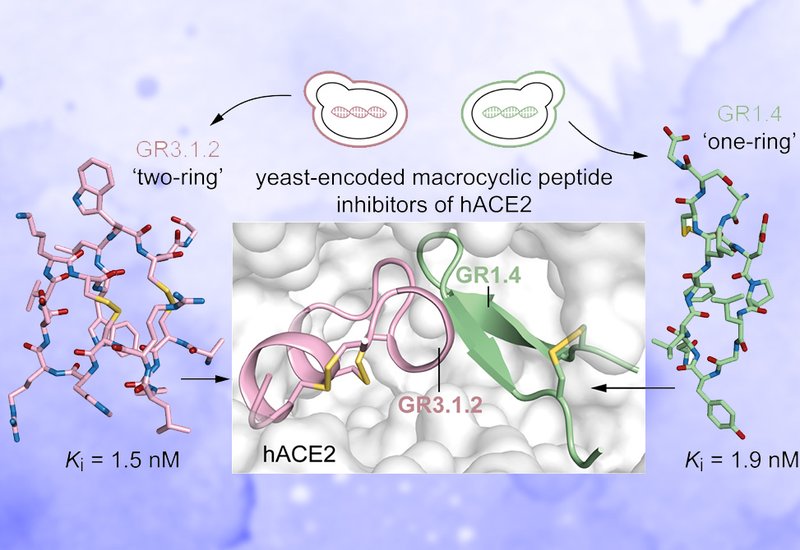
Sequencing revealed three families, GR1, GR2, and GR3, with GR1 comprising roughly 83% of selected clones. The GR1 family encodes 9-amino acid "one-ring" peptides sharing a conserved I/LGF/YN turn motif, notably lacking proline residues that characterize natural hACE2 substrates. Yeast surface titrations placed apparent KD values for GR1.1 and GR1.4 at 26.1 and 16.1 nM, respectively, and for the "two-ring" GR3.1 at 41.3 nM. All three ligands showed no detectable binding to eight unrelated proteins at 1 μM. Competition assays confirmed binding at the hACE2 catalytic pocket rather than at the receptor-binding domain site used by the SARS-CoV-2 spike protein. After chemical synthesis via Fmoc SPPS and orthogonal disulfide cyclization, enzyme inhibition assays delivered Ki values of 2.4 nM for GR1.1, 1.9 nM for GR1.4, and 1.5 nM for GR3.1.2, the correct disulfide isomer of the "two-ring" series. All three peptides showed no inhibition of the structurally related hACE1 at concentrations up to 100 μM. Human plasma half-lives measured at 37 °C by LC-MS reached 5.4 h for GR1.1, 6.5 h for GR1.4, and 8.6 h for GR3.1.2, compared to 24 min for a linearized serine-substituted analogue of GR1.4.
X-ray crystallography of the two lead inhibitors in complex with hACE2 resolved the structural basis for their activity. GR1.4 binds the active site at 2.39 Å resolution (PDB: 9RVT) as a rigid β-hairpin with two antiparallel β-strands bridged by a disulfide, burying 912 Å2 of surface. Alanine scanning confirmed that three of the four conserved turn residues are essential for binding. GR3.1.2 resolves at 2.02 Å (PDB: 28KD) in an unexpected cysteine-stabilized α-helix/α-helix fold resembling plant α-hairpinins, burying 1351 Å2 through an extensive network of polar contacts anchored by two arginine residues and multiple π-stacking interactions. Structural comparison with phage-display-derived BCY15291 and BCY15292 and mRNA-display-derived peptides 1, 2, and 6 showed that the yeast-derived macrocycles occupy lateral and opposing subregions of the catalytic pocket not contacted by the earlier inhibitors, with fewer than 20% of contacting hACE2 residues shared between the two sets.
The study demonstrates that yeast display can reach low-nanomolar inhibitory potencies against a therapeutically relevant metalloprotease starting from naïve combinatorial libraries at least 100-fold smaller than those used in phage and mRNA campaigns, without affinity maturation. The ability to characterize binding directly on the yeast surface compresses the hit-to-affinity timeline and provides continuous, quantitative visibility into the selection process. The discovery of distinct binding modes compared to previously reported inhibitors widens the structural space available to hACE2-targeted drug development, an advantage when lead optimization encounters dead ends with existing scaffolds. Two provisional patents and a spin-out company, Arzanya S.r.l., have already been established around this technology and the inhibitors described here, signaling a translational trajectory toward cardiovascular and inflammatory bowel disease therapeutics.