Nanopore sequencing revolutionized genomics by reading DNA molecules one base at a time. Extending this technology to peptides would transform proteomics, enabling single-molecule detection of protein variants and post-translational modifications that mass spectrometry struggles to resolve. The challenge lies in fundamental differences between nucleic acids and peptides. DNA carries uniform negative charge and predictable structure, properties that drive reliable capture and controlled movement through nanopores. Peptides offer no such consistency. Their charges vary from positive to negative, their lengths span wide ranges, and they lack the structural regularity that makes DNA so amenable to sequencing.
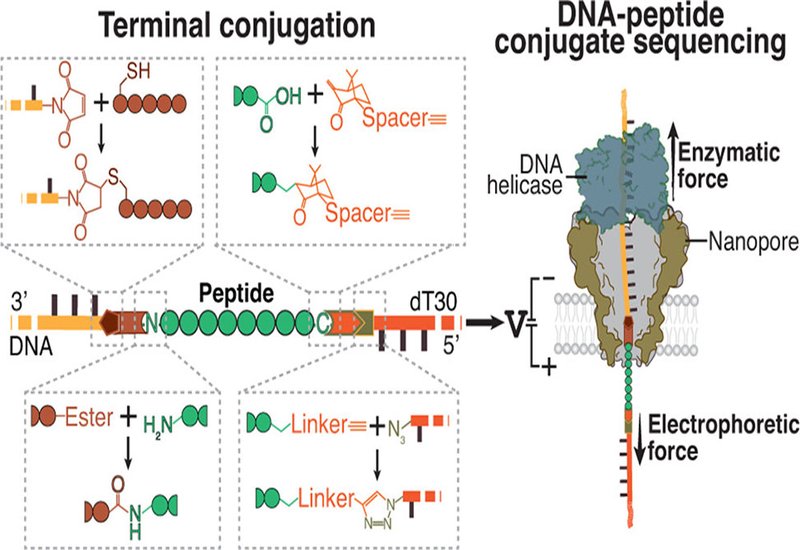
Researchers in the Cees Dekker Lab at Delft University of Technology, in collaboration with Bauke Albada at Wageningen University, published in the Journal of the American Chemical Society, have developed conjugation strategies that adapt native peptides for motor-driven nanopore sequencing. Their approach attaches DNA handles to peptide termini, allowing helicase enzymes to control translocation speed and direction. The DNA provides the motor engagement while the attached peptide generates sequence-dependent ionic current signatures as it passes through the nanopore constriction.
At the N terminus, the team employed omniligase to catalyze peptide bond formation between a DNA-linked acyl donor and target peptides. This enzymatic reaction proceeds within minutes at room temperature and tolerates diverse sequences. At the C terminus, they used photoredox-mediated decarboxylation to activate the terminal carboxylate under visible light, installing an alkyne handle for subsequent copper-catalyzed azide-alkyne cycloaddition to azide-functionalized DNA. Neither chemistry requires pre-installed reactive groups or engineered recognition motifs, making the approach compatible with native peptides from biological samples.
Testing eleven peptides ranging from 8 to 26 residues revealed clear design rules. Long peptides carrying net negative charge produced reproducible ionic current patterns with single-end conjugation alone. The 3X FLAG peptide and synthetic sequences containing glutamate residues generated discriminative signatures that enabled variant identification even among closely related sequences. Short peptides failed under these conditions, producing inconsistent signals and premature event termination. The team traced this failure to insufficient electrophoretic stretching force, which left short sequences vulnerable to thermal fluctuations and surface interactions within the pore.
Dual-terminal conjugation solved the short peptide problem. Adding a negatively charged DNA threading tail to the C terminus stabilized capture and maintained tension across the molecule during translocation. Both 1X FLAG and β-amyloid fragments that previously yielded unreadable traces now generated distinct signatures. Positively charged peptides presented a different challenge. Their cationic residues experience electrophoretic force opposing the direction of DNA-driven translocation, creating molecular tug-of-war that prevents controlled movement. Chemical acetylation of lysine side chains neutralized this positive charge, restoring successful sequencing of previously intractable histone-derived peptides.
The conjugation chemistries operate under mild, orthogonal conditions and proceed in aqueous solution. Omniligase ligation completes in five minutes, making the workflow compatible with temperature-sensitive samples. The photoredox step requires only visible light and a flavin photocatalyst. Together, these methods establish the first general approach for adapting unmodified peptides to nanopore sequencing, moving the technology beyond synthetic model systems toward authentic proteomic applications. Single-molecule protein analysis, long a distant goal, now has a chemical foundation.