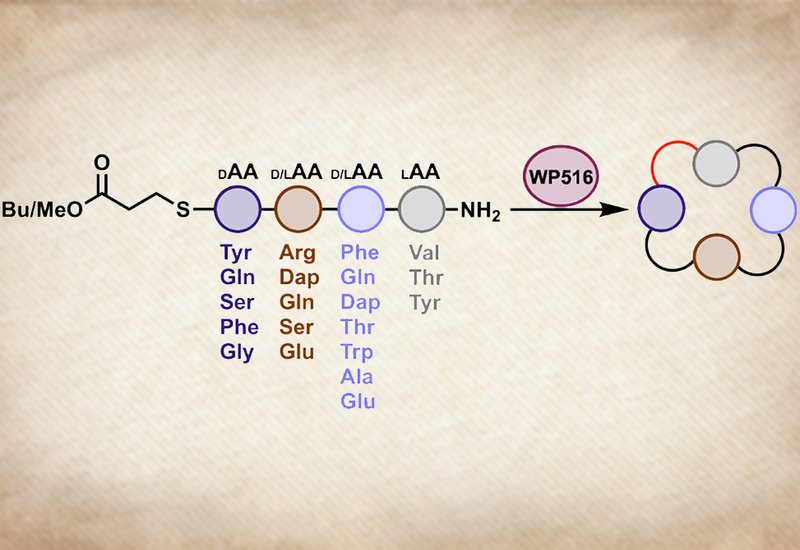
Cyclic tetrapeptides occupy a tantalizing corner of chemical space. Head-to-tail macrocyclization eliminates the charged termini that limit membrane permeability, while the constrained ring enforces secondary structures that boost potency, selectivity, and resistance to proteolytic degradation. Natural cyclic tetrapeptides, CTPs, already inhibit histone deacetylases, block calcium channels, and antagonize opioid receptors. Yet making them synthetically remains a persistent headache. The ground-state geometry of the peptide bond fights the conformation needed for ring closure, transannular strain punishes small rings, and competing oligomerization and C-terminal epimerization erode yields. Existing chemical strategies often demand specific residues to coax the backbone into a cyclization-friendly shape, severely restricting the sequences that chemists can access.
Researchers supervised by Professor Elizabeth I. Parkinson at Purdue University, published in Biochemistry, hypothesized that nature had already solved this problem in unexplored biosynthetic gene clusters. Penicillin-binding protein-type thioesterases, PBP-TEs, act independently of the nonribosomal peptide synthetase assembly line and consistently catalyze head-to-tail cyclization with unusual substrate tolerance. The team used bioinformatics to scan for PBP-TEs encoded alongside cryptic NRPS gene clusters containing exactly four adenylation domains, reasoning that these enzymes would natively cyclize tetrapeptides. From ten candidates, they expressed and characterized WP516 from Nonomuraea candida HMC10, the first PBP-TE discovered entirely through genome mining from a cryptic cluster with no associated known natural product.
WP516 proved to be a remarkably versatile tetrapeptide cyclase. The enzyme efficiently cyclized substrates across all four heterochiral stereochemical configurations, DLDL, DDDL, DDLL, and DLLL, a dramatic improvement over the only other reported tetrapeptide cyclase Ulm16, which fails entirely on DLLL peptides. WP516 tolerated a wide range of amino acid substitutions at most ring positions and accepted a C-terminal glycine, a residue that stymies most PBP-TEs without engineering. On a preparative 44-milligram scale, just 0.013 mol% catalyst achieved full conversion in two hours, delivering the purified cyclic product in 50% isolated yield, the first demonstration of any PBP-TE producing cyclic peptide on an isolable scale. Computational modeling with AlphaFold3, covalent docking, and molecular dynamics simulations revealed that a single residue swap in the α/β-hydrolase domain, tryptophan at position 300 in WP516 versus threonine at the equivalent position in Ulm16, plays a central role in determining tetrapeptide substrate tolerance. Introducing this substitution into Ulm16 as a T304W point mutant boosted its DLDL cycloselectivity roughly tenfold and enabled DLLL tetrapeptide cyclization for the first time.
This work establishes a generalizable bioinformatics workflow for mining cryptic gene clusters to find new peptide cyclases, sidestepping the traditional requirement for a known natural product. WP516 substantially expands the biocatalytic toolbox for CTP production, and the identification of a single residue governing ring-size preference provides a clear foothold for engineering even broader substrate scope. For a class of molecules with compelling therapeutic properties but limited synthetic accessibility, biocatalytic macrocyclization through enzymes like WP516 offers a practical path forward.