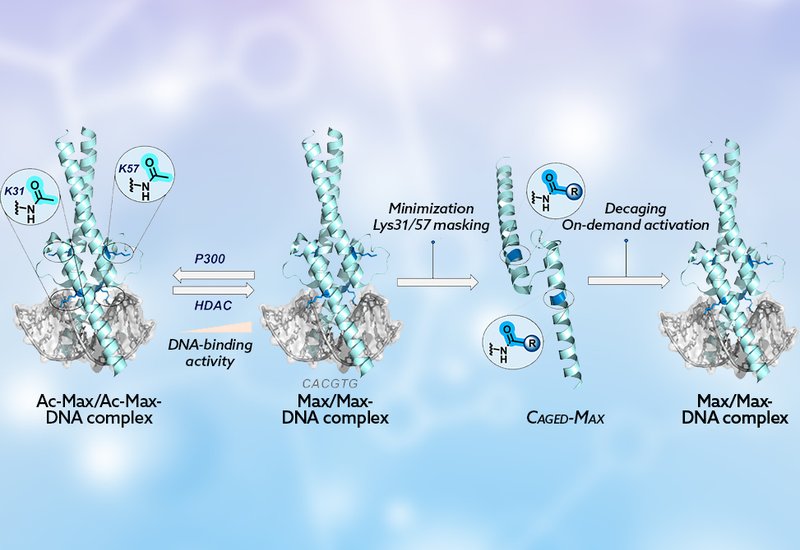
Transcription factors, TFs, govern gene expression by recognizing specific DNA sequences, and their activity is tightly regulated by post-translational modifications, PTMs, such as phosphorylation and acetylation. Among the roughly 1,600 TFs encoded in the human genome, Max occupies a pivotal position: as the obligate partner of Myc, it controls approximately 15% of all human genes, and disruption of the Myc–Max–Mad regulatory network is implicated in nearly 70% of human cancers. Engineering TFs that respond to external stimuli would offer a powerful handle for dissecting gene regulation and, ultimately, for therapeutic intervention, yet generating site-specifically caged full-length TF proteins using conventional biological methods has remained a stubborn challenge.
Researchers in the Jbara Group at Tel Aviv University, published in Bioconjugate Chemistry, addressed this challenge by designing a synthetic, light-activatable variant of the Max DNA-binding domain. Their earlier chemical synthesis work had revealed that acetylation at Lys31 and Lys57 disrupts critical salt bridges between Max and the phosphate-diester backbone, silencing DNA recognition. Building on that insight, the team masked those same lysine residues with the o-nitroveratryloxycarbonyl, Nvoc, photocaging group to create Max-Nvoc, a TF that is dark-stable and functionally dormant until illuminated.
The synthetic route itself required creative problem-solving. An initial native chemical ligation, NCL, desulfurization sequence failed because the Nvoc group decomposed under radical desulfurization conditions. The team pivoted to a one-pot NCL strategy coupled with palladium-mediated C–S cross-coupling, specifically an S-arylation step using a Pd(II) oxidative addition complex bearing a phenyl residue, to convert a cysteine at the ligation junction into a phenylalanine mimic without disturbing the photocaging groups. This three-step sequence delivered Max-Nvoc in 29% overall yield after reversed-phase HPLC purification, and LC–MS confirmed the product mass of 10,118.8 ± 1.3 Da against a calculated value of 10,117.2 Da.
Functional characterization confirmed that caging was highly effective. Electrophoretic mobility-shift assay, EMSA, experiments showed that Max-Nvoc, whether as a homodimer or as a heterodimer with a TAMRA-labeled Myc fragment, displayed dramatically reduced binding to the canonical enhancer box, E-box, DNA sequence even at elevated protein concentrations. Biolayer interferometry quantified the cost of caging precisely: Max-Nvoc bound E-box DNA with a KD of 161 μM, a roughly 12,000-fold weaker affinity than the 13 nM KD measured after photolytic removal of the Nvoc groups. UV irradiation at 350 nm in aqueous buffer restored binding with 99% decaging conversion within 60 minutes, and in situ irradiation of Max-Nvoc preincubated with DNA produced a progressive, time-dependent restoration of E-box binding detectable by EMSA. Circular dichroism spectroscopy confirmed that both the caged and decaged forms adopt the expected α-helical fold, establishing that the Nvoc groups suppress DNA binding through steric and electrostatic interference at the protein–DNA interface rather than by perturbing secondary structure.
The work demonstrates that total chemical protein synthesis, when combined with orthogonal late-stage transformations, can access photoreactive proteins that are inaccessible by biological expression or by classical ligation–desulfurization routes. The one-pot NCL–S-arylation strategy is broadly applicable and should extend to other desulfurization-incompatible chemical modifications. Looking ahead, the authors note that translating this optochemical control into living cells will require next-generation photolabile groups that respond to longer, tissue-penetrant wavelengths, a direction already under active investigation in their laboratory. The caged Max platform offers a template for building light-gated synthetic TFs targeting other oncogenic transcriptional circuits.