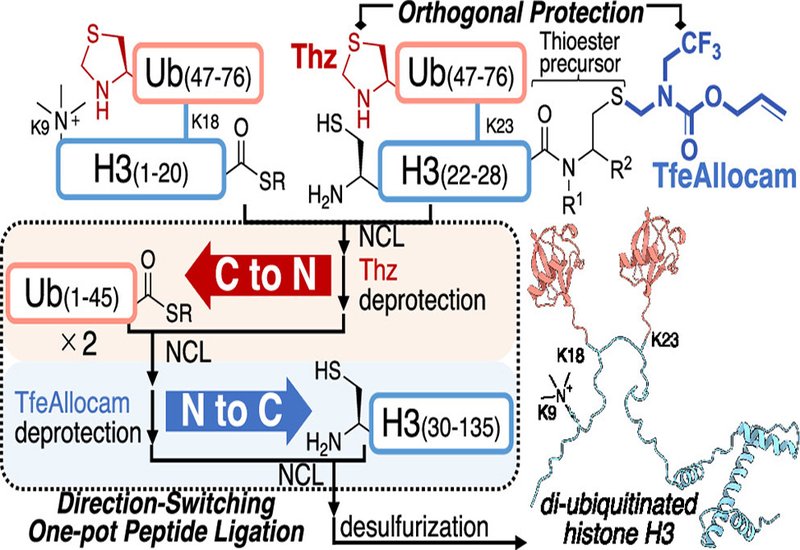
Chemical protein synthesis enables researchers to build proteins carrying modifications that biological expression systems cannot install, from site-specific ubiquitination to trimethylated lysine residues. The core chemistry relies on native chemical ligation, where a C-terminal thioester reacts with an N-terminal cysteine to form a native peptide bond. When assembling a large protein from multiple segments, chemists must ligate these pieces sequentially in a single reaction vessel to avoid the yield losses that come with repeated purification. Existing one-pot strategies work in a single direction, either building from C-terminus to N-terminus or from N-terminus to C-terminus. However, intermediates formed during ligation can aggregate unpredictably depending on the assembly order, and a scheme that works in one direction may fail in the other. Until now, no method has allowed chemists to switch ligation direction mid-synthesis within a single pot, limiting their ability to optimize assembly routes for difficult target proteins.
Researchers in the Hayashi, Murakami, and Okamoto Groups at Nagoya University and the University of Tokyo, published in the Journal of the American Chemical Society, developed the first direction-switching one-pot peptide ligation strategy. Their approach pairs two orthogonal protecting groups on cysteine residues: the established thiazolidine group for C-to-N ligation and a newly designed allylic thiol protecting group called TfeAllocam for N-to-C ligation. TfeAllocam incorporates a trifluoroethyl electron-withdrawing substituent that solves a long-standing stability problem with earlier Allocam protecting groups, which degraded under the acidic and basic conditions routinely used in solid-phase peptide synthesis. The team systematically compared TfeAllocam against two other new Allocam derivatives and demonstrated that it survives standard synthesis conditions with greater than 98% integrity while remaining quantitatively removable by palladium or ruthenium catalysts within minutes.
A critical challenge was managing formaldehyde released during TfeAllocam removal, which would cap free N-terminal cysteines as unreactive thiazolidines. The team solved this by deploying aminobenzamide-based formaldehyde scavengers that trap the aldehyde through rapid ring-closing reactions without interfering with ligation chemistry. With both protecting groups and their deprotection conditions fully orthogonal, the researchers demonstrated that the same set of four peptide segments could be assembled through either of two direction-switching routes, C-to-N then N-to-C or the reverse, simply by changing the order of deprotection steps. Both schemes delivered the target polypeptide cleanly in 34 to 41% isolated yield. To showcase the method on a demanding synthetic target, the team accomplished the semisynthesis of dimonoubiquitinated histone H3 bearing K9 trimethylation, a 287-amino acid protein carrying three distinct post-translational modifications critical for DNA methylation maintenance. Five peptide segments were condensed through three sequential ligations and two deprotection steps, all in a single reaction vessel completed within two days, yielding the full-length modified histone at 13% overall yield after desulfurization and purification. The synthetic protein was successfully incorporated into a reconstituted nucleosome.
This direction-switching strategy fundamentally expands the toolkit for chemical protein synthesis by allowing researchers to test alternative assembly routes from identical starting materials without resynthesizing peptide segments. The TfeAllocam and DaoAllocam protecting groups introduced here also have broader potential applications in the synthesis of disulfide-rich peptides, multicysteine cyclic peptides, and biological decaging reactions. As the field pursues increasingly complex modified proteins for structural biology and drug discovery, the ability to flexibly optimize ligation schemes within a single pot addresses a practical bottleneck that has constrained the ambition of synthetic protein chemists.