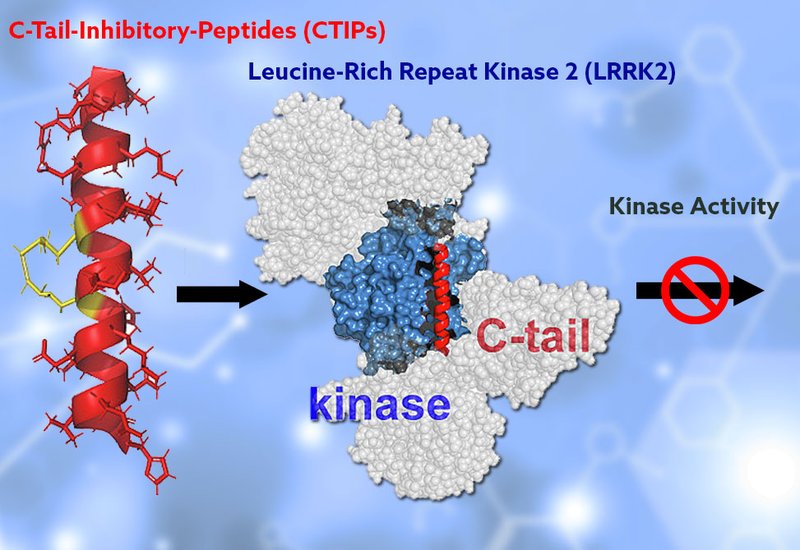
Parkinson's disease affects more than 10 million people worldwide and carries no cure. Among its genetic drivers, leucine-rich repeat kinase 2, LRRK2, stands out as the most frequently mutated gene in familial cases, and its variants also contribute to sporadic disease. Pathogenic mutations elevate LRRK2 kinase activity, which in turn disrupts downstream Rab GTPase signaling and drives hallmark cellular phenotypes including centrosomal cohesion defects and impaired ciliogenesis. ATP-competitive type I inhibitors can suppress this aberrant activity, but they stabilize a closed kinase conformation that causes LRRK2 to mislocalize into filamentous, microtubule-associated structures, an effect linked to lung and kidney toxicities observed in preclinical models. Alternative strategies that inhibit pathogenic LRRK2 activity without perturbing its physiological distribution remain an open challenge.
Researchers in the Kennedy Group at the University of North Carolina at Chapel Hill, published in Bioorganic Chemistry, set out to exploit a structural feature of LRRK2 that had been overlooked as a drug target. Cryo-EM structures of full-length monomeric LRRK2 reveal that a C-terminal helix leans against the back of the kinase domain and contacts the ankyrin and C-terminal of ROC domains, suggesting a scaffolding role that may regulate kinase activity through intramolecular domain-domain interactions. The team designed a focused library of 24-residue hydrocarbon-stapled peptides, termed C-terminal helix Inhibitory Peptides, CTIPs, derived from this helix sequence. Staples were formed by incorporating (S)-N-Fmoc-2-(4-pentenyl)alanine at i and i+4 positions and cyclizing via ring-closing metathesis, constraining the peptides into stable α-helical conformations. Methionine residues were replaced with norleucine to improve oxidative stability, and an N-terminal PEG3 linker was added to improve hydrophilicity. Peptides were assembled by Fmoc-based SPPS on Rink amide MBHA resin and purified by RP-HPLC.
Circular dichroism spectroscopy confirmed that CTIP3 and CTIP4, the two lead candidates, display characteristic α-helical minima at 208 and 222 nm, while their unstapled counterpart shows a random-coil signature. Fluorescence microscopy and flow cytometry in RAW264.7 macrophages, which express high endogenous LRRK2 levels, demonstrated efficient intracellular accumulation of FAM-labeled CTIPs at 10 μM. Both peptides reduced phosphorylation of the LRRK2 substrates Rab10 at T73 and Rab12 at S106 by up to 65% across a 1 to 5 μM concentration range in RAW264.7 cells, with up to 90% inhibition of Rab10 phosphorylation in HEK293 cells. Scrambled control peptides, CTIPScr3 and CTIPScr4, produced no measurable reduction in substrate phosphorylation, establishing sequence-dependent inhibition. Streptavidin pull-down assays using biotinylated CTIPs confirmed direct engagement of endogenous LRRK2 in RAW264.7, A549, and HEK293 cell lysates, while scrambled controls failed to enrich the kinase.
Microscale thermophoresis with purified full-length LRRK2 yielded dissociation constants of 5.5 μM for CTIP3 and 10.3 μM for CTIP4, providing quantitative, cell-free binding evidence. Western blot analysis showed no reduction in LRRK2 phosphorylation at Ser935 upon CTIP treatment, a site whose dephosphorylation is a hallmark of type I inhibitor engagement. GFP-tagged LRRK2 maintained diffuse cytosolic distribution in CTIP-treated HEK293 and RAW264.7 cells, in contrast to the filamentous relocalization induced by MLi-2. In LRRK2 R1441C knock-in mouse embryonic fibroblasts, both CTIPs restored centrosomal cohesion at 5 and 10 μM within 4 hours, and rescued ciliogenesis at 2.5 μM under serum-free conditions, achieving rescue levels comparable to MLi-2. Neither peptide affected LRRK2 protein levels or cell viability over treatment periods of up to 72 hours.
These findings position the LRRK2 C-terminal helix as a regulatory element amenable to allosteric peptide intervention and introduce CTIPs as the first peptide-based modulators of this interface. Because the C-terminal helix sequence diverges considerably from that of the closest LRRK2 homolog, LRRK1, and from other kinases, a degree of selectivity appears plausible, though kinome-wide profiling will be needed to confirm specificity. CTIPs are not expected to cross the blood-brain barrier in their current form, but the authors note that sequence modifications incorporating cell-penetrating motifs such as penetratin or RVG-29, or encapsulation in nanoparticles or extracellular vesicles, could extend CNS access. Peripheral tissues and immune cells also express LRRK2 at high levels and are relevant to PD pathology as well as Crohn's disease, providing additional therapeutic contexts that do not require central nervous system delivery. Future structural studies of LRRK2 bound to CTIPs should clarify the precise allosteric mechanism and guide next-generation optimization.