Bacterial RNA polymerase, RNAP, is essential to gene expression in all bacteria and a proven target for broad-spectrum antibiotics. Pseudouridimycin, PUM, is a naturally occurring C-nucleoside/peptide antibiotic that inhibits RNAP by engaging residues directly in the catalytic site, yielding a spontaneous resistance rate an order of magnitude lower than existing RNAP-targeted drugs. PUM is active against drug-resistant and multidrug-resistant strains of Gram-positive and some Gram-negative pathogens. One fundamental obstacle has blocked its development: the central hydroxamate bond undergoes self-immolative cleavage at physiological pH, rendering PUM chemically unstable before it can reach its target.
Researchers in the Del Valle Group at the University of Notre Dame and the Ebright Group at Rutgers University, published in ACS Medicinal Chemistry Letters, combined cryo-electron microscopy with solid-phase peptide synthesis, SPPS, to characterize a stabilized des-hydroxy PUM analog and guide structure-activity relationship, SAR, studies across 50 new compounds. In the des-hydroxy analog 2a, the labile hydroxamate is replaced by a secondary amide, restoring chemical stability while retaining low-micromolar RNAP-inhibitory activity. The team determined cryo-EM structures of native PUM and 2a bound to an Escherichia coli RNAP transcription complex, providing a more clinically relevant structural reference than the Thermus thermophilus crystal structures reported previously.
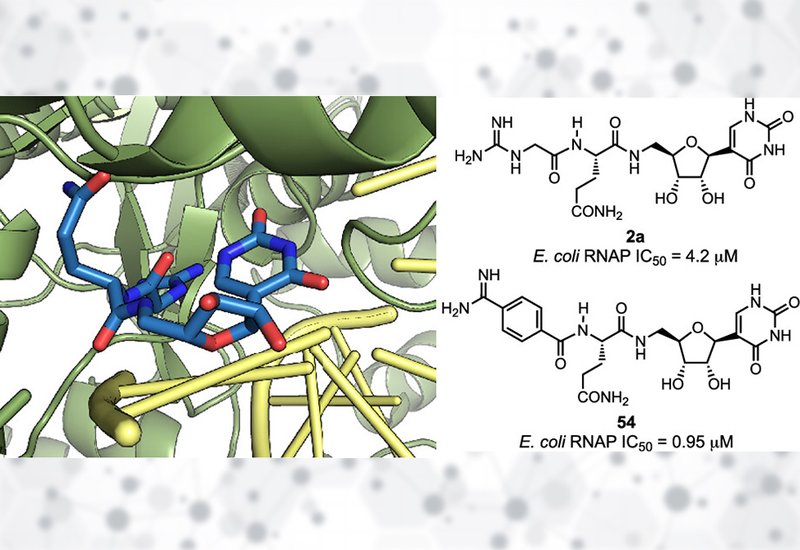
Both compounds bind the E. coli RNAP transcription complex in the trans geometry, resolving uncertainty from the earlier T. thermophilus crystal structure that showed cis geometry and confirming that the secondary amide in 2a is compatible with productive RNAP binding. In the trans conformation, the Gdn-Gly carbonyl oxygen hydrogen-bonds with the pseudouracil base in an interaction compatible with pseudouracil but not uracil, while the hGln carbonyl oxygen hydrogen-bonds with the 3′-hydroxyl of the nascent RNA. Native PUM forms one additional contact absent in 2a: the hydroxamate OH hydrogen-bonds with RNAP β subunit Lys1073, consistent with the modestly reduced potency of 2a relative to PUM.
The SPPS approach anchored a protected pseudouridyl amine onto a backbone-anchoring linker, BAL, resin, enabling parallel synthesis of multiple analogs within a single day. The first series explored Gln residue substitutions: the Tyr analog 6 was the most potent with an IC50 of 7.2 μM, likely through a π-π stacking interaction with RNAP β subunit His936, while substituting Asn for Gln caused a complete loss of activity despite being Gln's lower homologue. The Gdn-Gly tail proved less tolerant: methylation, homologation, truncation, or Cα substitution of the guanidine group all significantly reduced potency. A para-substituted phenyl-guanidine, compound 37, maintained activity comparable to 2a, and the corresponding para-substituted phenyl-amidine, compound 54, achieved an IC50 of 0.95 μM, a fourfold improvement and the most potent RNAP inhibitor in the series. Substituent regiochemistry proved critical: meta-positioned groups consistently abolished activity.
Eight analogs with IC50 values below 20 μM were evaluated for bacterial growth inhibition against Streptococcus pyogenes and Staphylococcus aureus. Only compound 37 matched the antibacterial potency of 2a against S. pyogenes. Despite its submicromolar RNAP-inhibitory activity, compound 54 showed no meaningful antibacterial activity against either species, suggesting the guanidine group plays a role in cellular entry beyond RNAP engagement. Stability studies in 25% human serum at 37 °C confirmed that native PUM degrades rapidly, whereas des-hydroxy analogs 2a, 37, and 54 remained completely stable for at least 72 hours.
These findings establish the trans-geometry E. coli RNAP complex as the appropriate structural framework for PUM analog design, identify the para-phenyl amidine scaffold as a submicromolar RNAP inhibitor with complete serum stability, and clarify the dual role of the guanidine group in enzymatic inhibition and cellular uptake. The SPPS platform developed here enables rapid access to further stabilized PUM derivatives and provides a concrete foundation for developing RNAP-targeted therapeutics against resistant bacterial pathogens.