Chemical protein synthesis offers access to proteins with precise sequences, modifications, and structures that biological expression cannot readily provide. The workhorse of the field, native chemical ligation, NCL, joins unprotected peptide fragments via a chemoselective reaction between a C-terminal thioester and an N-terminal cysteine to form a native amide bond. Its central limitation is the low natural abundance of cysteine, approximately 1.7% in natural proteins, which restricts where fragment junctions can be placed. Ligation-desulfurization strategies and thiol-auxiliary approaches have expanded the accessible sites, but all existing methods remain constrained to specific amino acid residues at the ligation junction, complicating the synthesis of targets where convenient residues are absent or poorly spaced.
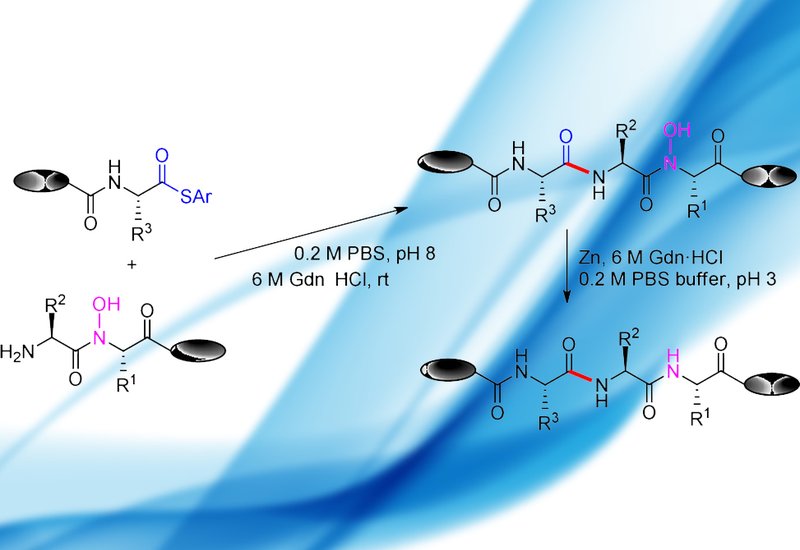
Researchers in the Fu Group at Tsinghua University in Beijing, published in Organic letters, have developed a new chemoselective peptide ligation strategy, hydroxylamine-involved ligation, HAL, that operates without dependence on any specific amino acid at the junction site. The approach couples C-terminal peptide thioesters with N-terminal aminoacyl-N-hydroxy peptides in aqueous buffer at pH 8 and room temperature. Building blocks carrying the N-hydroxyl group were prepared for all twenty proteinogenic amino acids and incorporated directly into solid-phase peptide synthesis, SPPS, without interfering with standard Fmoc coupling chemistry.
The reaction proceeds through two sequential steps. First, S,O-ester exchange between the peptide thioester and the N-hydroxyl group of the partner peptide forms an O-acyl isopeptide intermediate. Second, an O,N-acyl transfer across a geometrically favorable six-membered ring yields the ligated N-hydroxyl peptide. Zinc reduction in aqueous medium at pH 3 then converts the N-hydroxyl bond to a native amide, completing the ligation. Optimized at pH 8 and room temperature, the reaction reached completion within six hours, with ligated N-hydroxyl peptides isolated in 54 to 78% yields across a 25-compound substrate scope and final reduced peptides obtained in 73 to 96% yields. Critically, the method accommodates sterically demanding junction combinations including valine-valine, valine-threonine, and valine-proline pairings that are notoriously problematic for existing ligation strategies. Minor epimerization at the C-terminal residue of the thioester fragment was observed and attributed to a six-membered cyclic intermediate; epimers were separable by preparative HPLC. The strategy was validated in the total synthesis of ubiquitin, a 76-residue protein assembled in the N-to-C direction from three fragments, with an overall yield of 28% and the target protein confirmed by ESI-MS.
HAL adds a genuinely site-independent tool to the chemical protein synthesis repertoire, complementing NCL and its derivatives rather than replacing them. By removing the constraint of specific junction residues, it broadens the range of retrosynthetic disconnections available to protein chemists and should prove especially useful for targets where cysteine or other ligation-compatible residues are absent at convenient positions. The aqueous, room-temperature conditions and compatibility with standard SPPS building blocks lower the barrier to adoption, and the demonstrated tolerance for challenging steric combinations suggests the strategy will extend to substrates that have resisted ligation by existing methods.