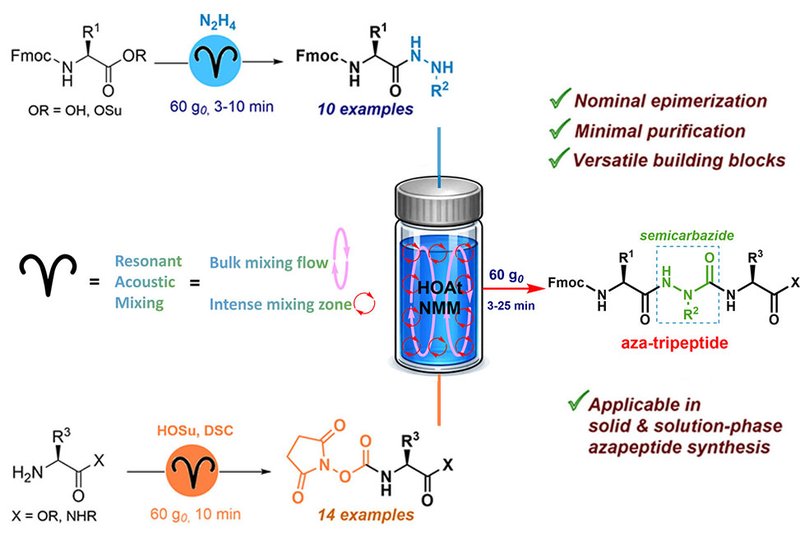
Azapeptides, in which a semicarbazide unit replaces one or more backbone amide bonds, have attracted growing interest in therapeutic development because the substitution boosts metabolic stability and can sharpen biological activity. Several clinical candidates owe their durability to exactly this modification. Yet the chemistry required to build azapeptides reliably at scale has remained stubbornly difficult. Selective functionalization of hydrazine intermediates, amino acylation of the poorly nucleophilic semicarbazide, unwanted hydantoin ring closure, epimerization at sensitive stereocenters, and the need for chromatographic cleanup at multiple stages have all conspired to keep azapeptides expensive and difficult to access in useful quantities.
Researchers in the Lubell Lab at Université de Montréal, published in Organic Letters, addressed these obstacles by designing a solution-phase route built around resonant acoustic mixing, RAM, a solvent-lean mechanochemical technique that uses a balance of low-frequency acoustic and vibrational energy to drive reactions at high concentration. The core innovation is a family of N-Fmoc-aza-tripeptide building blocks that consolidate the semicarbazide unit into a single, stable, pre-formed fragment, allowing one-step coupling onto a growing peptide chain during solid-phase synthesis.
The team first established clean access to N-Fmoc-amino hydrazides via hydrazinolysis of activated esters and EDCl-mediated couplings under RAM conditions. Critically, epimerization was undetectable at most positions by chiral SFC-MS analysis, and even the stereochemically demanding His residue could be handled cleanly when fresh N-hydroxysuccinimide esters were used. To generate the active carbamate partners, the researchers turned to N,N′-disuccinimidyl carbonate, DSC, in place of the more common p-nitrophenyl chloroformate. DSC suppressed urea byproduct formation, allowing succinimidyl carbamates to be isolated in 80–98% yields across a broad range of amino ester and amino amide substrates. The resulting carbamates proved shelf-stable for up to three months at −20 °C. Coupling hydrazide and active carbamate components in NMM with catalytic 1-hydroxy-7-azabenzotriazole, HOAt, under RAM then delivered aza-di-, -tri-, and -tetrapeptide intermediates in 65–84% yields, with excess carbamate conveniently quenched to a water-soluble urea by treatment with N,N-dimethylaminoethylamine. Using N-methylmorpholine, NMM, as solvent proved decisive: NMM’s exceptional solubilizing power dissolved otherwise intractable hydrazide components under conditions that suppressed dibenzofulvene formation from Fmoc cleavage.
Solid-phase synthesis demonstrated the utility of the approach in biologically relevant contexts. An aza-glycine variant of the antimicrobial peptide H-KWKWKpGKWKWK-NH2 was assembled in 97% purity by coupling the aza-tripeptide Fmoc-D-Pro-azaGly-Lys onto a Rink amide resin-supported sequence without detectable epimerization or truncation. A second target, the cardioprotective CD36 modulator CP-3(iv), an aza-analogue of growth hormone releasing peptide-6, GHRP-6, was obtained in 95% purity after RP-HPLC purification. Both examples confirmed that aza-tripeptide building blocks couple cleanly and match the efficiency of the established aza-dipeptide strategy while delivering an additional residue in a single loading step.
The work reframes azapeptide synthesis as a genuinely scalable endeavor. By pairing RAM-compatible reagent systems with a building-block strategy that sidesteps the most troublesome selectivity problems, the Lubell Lab has lowered the practical barrier to exploring semicarbazide-containing peptides in drug discovery programs. The route requires no specialist equipment beyond a RAM instrument, uses reduced solvent volumes, and avoids toxic reagents such as phosgene. As interest in metabolically stabilized peptide therapeutics continues to grow, this methodology offers a direct path from amino acid starting materials to complex azapeptide candidates with the stereochemical fidelity and purity profiles demanded by preclinical development.