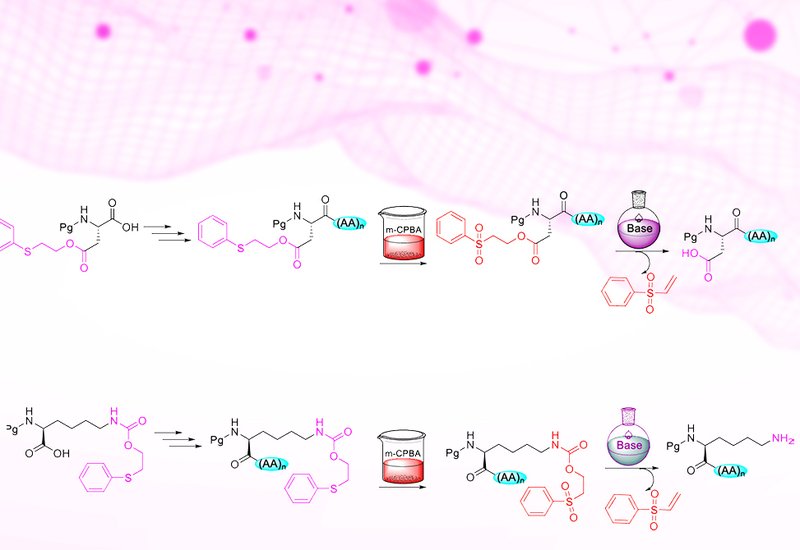
Modern peptide synthesis demands precise control over the functional groups of amino acids. The carboxyl, amine, hydroxyl, and thiol moieties that define amino acid chemistry are also the source of its complexity: without adequate protection, these groups react indiscriminately, producing side products, low yields, and stereochemical scrambling. Standard Fmoc/tBu solid-phase peptide synthesis addresses this challenge through orthogonal protecting groups removed by distinct chemical mechanisms, but the growing structural complexity of therapeutic peptides, including branched sequences, cyclic frameworks, and fatty acid conjugates, strains the limits of two-dimensional orthogonality. Adding a third dimension of protecting group selectivity typically requires introducing new reaction types, such as photolysis or palladium-catalyzed deprotection, that bring their own handling challenges and limit accessibility in general laboratory settings.
Researchers supervised by Professors Fernando Albericio and Beatriz G. de la Torre at the University of KwaZulu-Natal developed a new family of safety-catch protecting groups based on the phenylthioethyl, Pte, and phenylsulfonylethyl, Pse, moieties, published in Organic Letters. A safety-catch protecting group is one that resists removal under conditions that strip other protecting groups, but becomes labile after a simple chemical modification, allowing the same base chemistry to remove both. In this system, Pte esters and carbamates are stable to piperidine and trifluoroacetic acid, the two reagents that drive standard Fmoc/tBu synthesis. Oxidation of the thioether to a sulfone with m-CPBA converts the protecting group to its Pse form, which is then cleanly removed by base through a beta-elimination mechanism analogous to Fmoc removal. For carboxylic acid protection on glutamate and aspartate, piperidine or diethylamine suffices for Pse removal. For amine protection on the lysine side chain via the Pteoc carbamate, a solution of 5% DBU and 5% 4-methylpiperidine in DMF achieves complete deprotection in two 30-minute treatments.
The group validated Pte(oc) stability across a range of TFA concentrations and base treatments, confirming compatibility with the full suite of standard SPPS conditions. They then applied the protecting group to three increasingly complex synthetic challenges. First, on-resin homodetic cyclization of a model octapeptide was achieved by selectively unmasking the glutamate carboxyl and lysine amine and coupling them with PyOxim, with no interference from the rest of the protecting group scheme. Second, on-resin installation of fatty acid chains on the lysine side chain was demonstrated, including the complex AEEA-AEEA-gamma-Glu-eicosanoic acid linker used in GLP-1 receptor agonists such as tirzepatide. Third, a sequential multifunctionalization strategy placed four orthogonal lysine protecting groups, Fmoc, Mtt, Alloc, and Pteoc, on a single peptide chain and removed them in series to install four distinct amino acids at defined positions, confirming that Pteoc is fully stable to the conditions required to remove each of the other three groups.
This safety-catch approach eliminates the need for palladium-catalyzed deprotection in many synthetic contexts, lowering the technical barrier for laboratories without specialist expertise in transition metal chemistry. The result is a versatile and operationally practical strategy for the synthesis of branched, cyclic, and lipidated peptides of the kind increasingly prominent in pharmaceutical pipelines. Its compatibility with standard Fmoc/tBu chemistry positions Pte(oc) as a straightforward addition to the protecting group toolkit available to synthetic peptide chemists working across both research and industrial settings.