Engineering the translational machinery to incorporate backbone-modified monomers could enable the biosynthesis of sequence-defined polymers with new-to-nature properties. β-Hydroxy acids are particularly attractive because they could support the programmed production of biocompatible polyesters and natural product-like depsipeptides. Yet despite early promise, yields of proteins containing even a single β2-monomer remain frustratingly low and unpredictable. Previous work demonstrated that both enantiomers of β2-hydroxy-Nε-Boc-lysine, β2-OH-BocK, are substrates for the orthogonal M. alvi pyrrolysyl-tRNA synthetase, PylRS, and tRNA pair in vitro, but only the (R) enantiomer is incorporated into protein in vivo. The question remained: where in the translation pathway does stereochemistry matter?
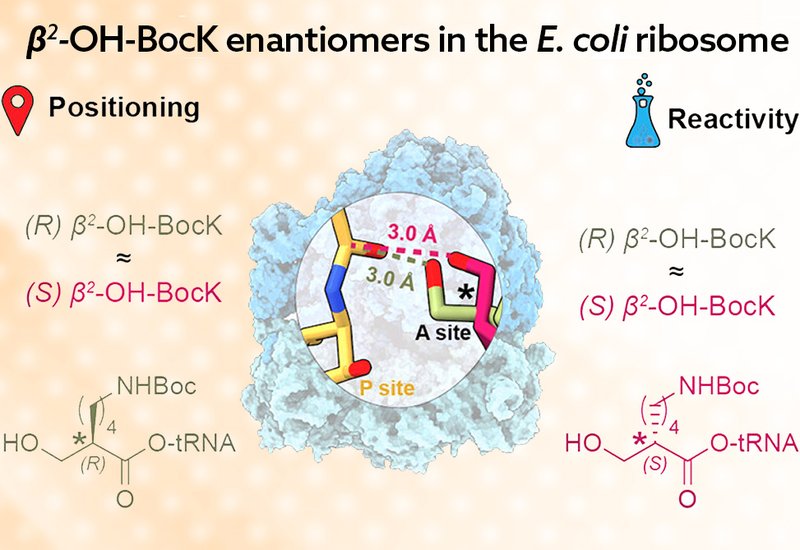
Researchers in the Cate and Schepartz Groups at the University of California, Berkeley, and the Lawrence Berkeley National Laboratory, published in the Journal of the American Chemical Society, used cryogenic electron microscopy to visualize how the E. coli ribosome accommodates tRNAs acylated with either the (R) or (S) enantiomer of β2-OH-BocK. They prepared ribosome complexes with formylmethionine-tRNA in the P site and β2-OH-BocK-tRNA in the A site, using 3′-amino-modified tRNAs to prevent hydrolysis during sample preparation. The resulting cryo-EM structures, refined to 2.14 and 2.29 Å resolution, provided unambiguous stereochemical assignment of each enantiomer within the peptidyl transferase center, PTC.
The structures revealed that despite their opposite stereochemistry, both β2-OH-BocK enantiomers are accommodated remarkably well within the ribosomal active site. The extended BocK side chains insert into the A-site cleft formed by nucleotides A2451 and C2452, just like the side chains of natural α-amino acids. Critically, both enantiomers induce the canonical induced-fit conformational change in the PTC, in which nucleotide U2506 rotates to confine the A-site side chain and U2585 moves away to expose the P-site carbonyl for nucleophilic attack. The hydroxyl nucleophiles of both enantiomers were positioned less than 3 Å from the P-site carbonyl carbon with Bürgi–Dunitz angles between 68 and 72 degrees, geometry that strongly favors bond formation. The ordered water network believed to facilitate proton transfers during catalysis was present in both complexes. The big surprise is that the additional methylene group defining the β2-linkage allows both enantiomers to rotate around the Cα–Cβ bond and position their nucleophilic oxygen atoms in nearly identical reactive positions, regardless of absolute stereochemistry.
To test whether structural equivalence translates to functional equivalence, the researchers performed in vitro translation experiments using purified components. They acylated tRNAPyl with (R) or (S) β2-OH-BocK using M. alvi PylRS, quantified acylation levels by mass spectrometry, and supplemented PURE translation systems with equivalent concentrations of each acyl-tRNA. Mass spectrometry analysis of FLAG-tagged peptide products showed incorporation ratios between 0.9 and 1.7, confirming no significant difference in how well the ribosome incorporates the two enantiomers. Luminescence assays using HiBiT peptide templates corroborate this finding. Together, the structural and biochemical data demonstrate that once delivered to the ribosome, both β2-OH-BocK enantiomers react with essentially equal efficiency. The substantial in vivo preference for the (R) enantiomer must therefore originate upstream of bond formation, likely at the level of tRNA acylation efficiency or delivery by EF-Tu. An accompanying study from the Chatterjee and Schepartz Groups shows that when cells are provided with an evolved aminoacyl-tRNA synthetase that efficiently charges tRNA with both enantiomers, both (R) and (S) β2-OH-BocK are incorporated into protein with high yield and fidelity, fully validating the predictions derived from these ribosome structures.