Ribosomally synthesized and post-translationally modified peptides, RiPPs, rank among the most structurally diverse families of natural products. Their biosynthesis begins with genetically encoded precursor peptides that undergo extensive enzymatic tailoring, generating scaffolds with broad-ranging bioactivities. Proteins bearing the conserved HEXXH motif constitute a large class of metalloproteases traditionally associated with peptide bond hydrolysis, yet recent studies have revealed that some HEXXH enzymes instead function as α-ketoglutarate-dependent, αKG-dependent, nonheme iron oxidases that carry out entirely different chemistry. Characterizing these unconventional enzymes promises to expand both the catalytic toolkit and the structural diversity accessible through RiPP biosynthesis.
Researchers in the van der Donk Group at the University of Illinois Urbana-Champaign, published in the Journal of the American Chemical Society, used genome mining to identify biosynthetic gene clusters from Pseudomonas strains encoding two HEXXH enzymes, PflC and PosC, alongside an unusual MNIO-nitroreductase fusion enzyme, PflD. The team heterologously expressed the precursor peptides with these tailoring enzymes in Escherichia coli, then characterized the products through high-resolution mass spectrometry, tandem MS, and extensive NMR analysis. They also reconstituted enzyme activity in vitro and conducted systematic mutagenesis to map the sequence requirements for each transformation.
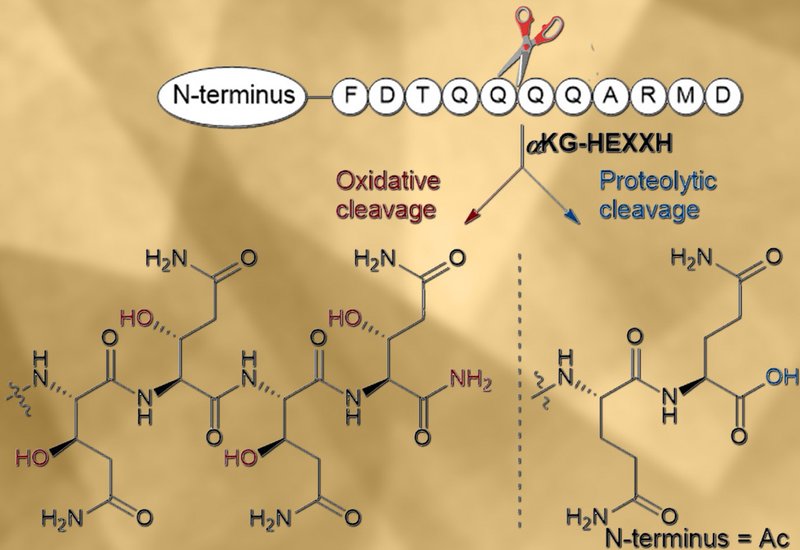
PflC and PosC each catalyzed two distinct reactions on the same substrate. Both enzymes hydroxylated the β-carbon of multiple consecutive glutamine residues and selectively recognized a conserved C-terminal ARMD tetrapeptide to trigger oxidative backbone cleavage, generating a terminal amide and releasing a ketone-containing byproduct. Mutational analysis revealed that the alanine at the first position of the ARMD motif served as the critical determinant for cleavage, while the remaining three residues proved individually dispensable. Remarkably, the ARMD motif did not need to occupy the C-terminus for scission to occur, as the enzyme consistently cleaved immediately upstream of the motif regardless of its position. When the researchers removed the N-terminal leader peptide and tested PflC against truncated synthetic substrates, the enzyme switched from oxidative cleavage to hydrolytic proteolysis at the same site, producing a carboxylic acid terminus rather than an amide. This finding demonstrated that leader peptide interactions directly modulate whether the enzyme activates oxygen for radical chemistry or instead acts as a conventional metalloprotease. The MNIO-nitroreductase fusion enzyme PflD contributed two additional modifications: its nitroreductase domain dehydrogenated a phenylalanine residue to produce a rare Z-dehydrophenylalanine, the first such transformation reported in RiPP biosynthesis, while its MNIO domain hydroxylated an aspartate residue at the β-carbon.
These findings reveal an unprecedented dual catalytic personality for αKG-dependent HEXXH enzymes, performing both side-chain hydroxylation and backbone scission within a single active site. The discovery that leader peptide binding gates the switch between oxidative and hydrolytic pathways provides a striking example of substrate-controlled reaction selectivity. Together with the novel dehydrophenylalanine formation by the nitroreductase domain, this work broadens the repertoire of post-translational modifications available in bacterial RiPP pathways and offers promising starting points for developing new biocatalysts.