Determining the stereochemistry of organic molecules demands two distinct analytical approaches: alignment media to measure anisotropic NMR parameters and chiral auxiliaries to differentiate enantiomers. Researchers typically run these as separate experiments using orthogonal reagents, adding time and complexity to postsynthesis characterization. Some lyotropic liquid crystals, LLCs, can partially bridge this gap, but most operate only in medium-polarity aprotic solvents and impose strong molecular ordering that leads to crowded spectra requiring two-dimensional analysis. A single medium capable of weak alignment and versatile chiral differentiation in polar, protic solvents has remained elusive.
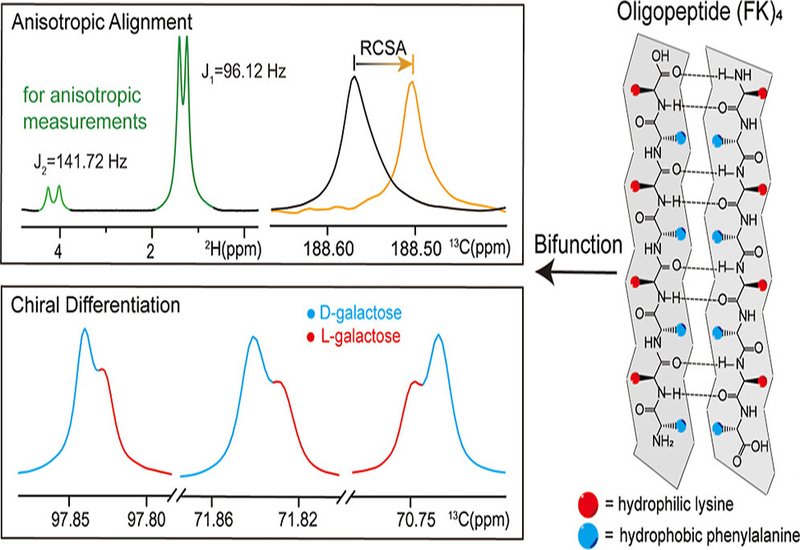
Researchers in the Liu Group at the Innovation Academy for Precision Measurement Science and Technology, Chinese Academy of Sciences, published in J. Am. Chem. Soc., demonstrate that the amphiphilic oligopeptide, phenylalanine-lysine tetrapeptide repeat, abbreviated (FK)4, self-assembles into β-sheet nanofibrils that simultaneously serve as an alignment medium and a chiral differentiating agent. The alternating hydrophobic phenylalanine and hydrophilic lysine residues follow a binary patterning that drives fibril formation. Transmission electron microscopy confirmed thin, rigid nanofibrils, and fast Fourier transform analysis revealed a meridional reflection near 4.7 Å, consistent with β-strand stacking. The resulting LLC phase weakly aligns solvent molecules while providing sufficient intermolecular contact with analytes for robust enantiomeric recognition across multiple nuclei and solvents.
The team tested (FK)4 across a chemically diverse analyte set using proton-decoupled 13C NMR spectroscopy. Every amino acid examined, including proline, glutamic acid, isoleucine, and phenylalanine, showed clear resonance bifurcation at one or more carbon sites, with peak integrations accurately matching prepared enantiomer ratios. Monosaccharides glucose, fructose, and galactose each exhibited chiral recognition at two to three carbon positions, as did the disaccharide lactose, pentose xylose, and tetrose threitol. Racemic pharmaceuticals including terbutaline, epinephrine, and chloroquine all produced resolvable signal separation. Enantiopurity quantification proved reliable: for leucine, integration of split 13C signals correlated with HPLC reference values at R2 averaging 0.961. High-sensitivity 1H NMR with (FK)4 resolved aspartic acid enantiomers into symmetric doublets suitable for direct ratio measurement, and 19F NMR differentiated 4-fluorophenyl ethylamine enantiomers with matching accuracy. Critically, (FK)4 remains NMR silent upon assembly, as aggregation-induced broadening suppresses oligopeptide signals and minimizes spectral interference. The medium functions in water, methanol, and isopropanol, and its anisotropic state switches reversibly to isotropic by brief thermal treatment, enabling residual chemical shift anisotropy, RCSA, measurements in the same sample. RCSA analysis of dihydrolycorine and α-santonin in methanol correctly assigned relative configurations for both compounds, confirming that (FK)4 operates as a reliable alignment medium beyond aqueous conditions.
The team applied both functions together in a streamlined postsynthesis workflow: a single (FK)4-containing sample first revealed enantiomeric composition via 13C signal bifurcation, then yielded relative configuration assignment after thermal-induced isotropic conversion and RCSA analysis, identifying the cross-coupling product as a racemic mixture of RS and SR 3-methyl-2-phenylvaleric acid. This dual-function approach compresses what typically requires separate experiments and reagent sets into a single analytical sequence. The authors note that an enduring limitation of RDC- and RCSA-based structural analysis has been the inability to differentiate enantiomers; (FK)4 directly addresses this gap and points toward future extension to absolute configuration elucidation.