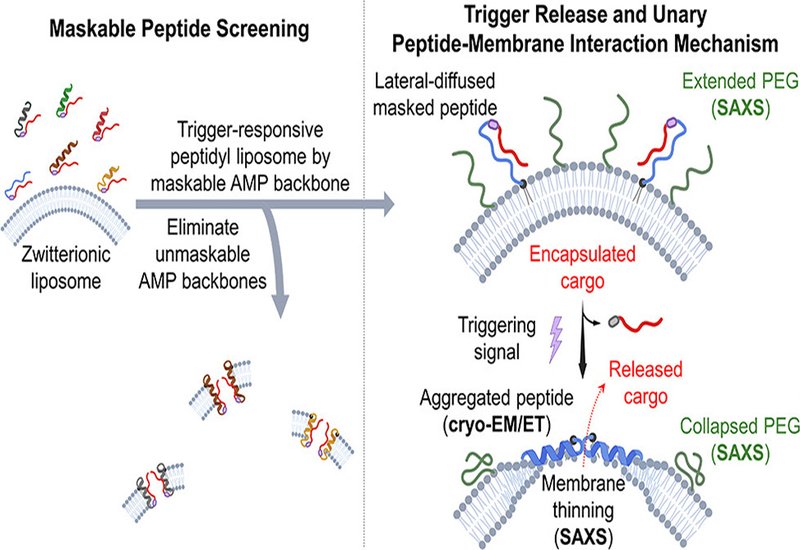
Liposomes are among the most clinically advanced drug delivery platforms, yet a fundamental tension persists between encapsulation stability and controlled release. Trigger-responsive peptidyl liposomes, in which antimicrobial peptides, AMPs, are conjugated directly to the liposome surface and activated on demand, offer a compelling solution, but every previous attempt to build such a unary system has been defeated by premature drug leakage after peptide conjugation. The underlying problem is that conjugation concentrates peptides at the membrane surface, drastically raising their local effective concentration and making membrane lysis far harder to suppress than in conventional binary peptide-membrane systems where peptide and liposome remain separate until mixing. Without knowing which AMP backbones can be reliably masked in this covalently tethered state, rational design has been impossible.
Researchers supervised by Professor Hsien-Ming Lee at the Institute of Chemistry, Academia Sinica, Taipei, Taiwan, published in J. Am. Chem. Soc., screened nine cationic amphipathic AMPs for maskability in a covalently conjugated unary configuration, using a covalent titration assay that quantifies the peptide-to-lipid ratio required for 50% liposomal release in both masked and unmasked states. The masking domain employed was a dodecaglutamate, 12E, that suppresses amphiphilic helix formation through intramolecular electrostatic interactions and impedes lateral peptide aggregation through intermolecular repulsion. A light-cleavable ortho-nitrobenzyl linker between the AMP and 12E allowed rapid, clean photounmasking without side reactions, enabling a controlled mechanistic study of peptide-membrane interactions.
Magainin 2, MAG2, emerged as the standout candidate by a wide margin, displaying a 34.8-fold increase in the peptide-to-lipid ratio required for release when masked, compared to 3.3- to 11.5-fold for the other candidates. Masked MAG2-conjugated liposomes loaded with doxorubicin showed negligible premature leakage across a range of temperatures and in serum-rich environments, while UV trigger-induced release reached approximately 80% within 30 to 60 minutes. Cell studies in KB cells confirmed that triggered masked MAG2 liposomes delivered doxorubicin efficiently to nuclei, with an IC50 of 2.0 μM closely matching free doxorubicin at 0.7 μM, representing a 46-fold inducible cytotoxicity increase over the untriggered state. Mechanistic characterization by cryo-EM, cryo-electron tomography, cryo-ET, small-angle X-ray scattering, SAXS, and circular dichroism, CD, revealed a previously uncharacterized sequence of structural events. In the signal-waiting state, freely laterally diffusing masked MAG2 suppresses amphiphilic folding and expands the outer PEG layer by approximately 21 Å through steric support. Upon photounmasking, MAG2 undergoes localized lateral aggregation, induces transient membrane defects in confined patches consistent with a carpet mechanism at low peptide-to-lipid ratios, and causes the PEG layer to recompact by approximately 16 Å as the freely diffusing peptides are immobilized into aggregates. Notably, MAG2, long considered inactive against zwitterionic membranes in binary systems, proved strongly membrane-lytic when surface-anchored, a proximity effect that also explains why this peptide class had previously been overlooked as a scaffold for liposomal delivery.
The work resolves a longstanding bottleneck in trigger-responsive drug delivery by identifying AMP backbone identity, rather than masking strategy alone, as the critical design variable in unary peptidyl liposome systems. The mechanistic framework it establishes, linking peptide secondary structure suppression, lateral diffusion, aggregation, and membrane remodeling into a coherent hierarchical sequence, provides generalizable design principles for next-generation peptide-engineered liposomes. The light trigger used here can in principle be replaced with disease-associated biochemical cues, opening a path toward clinically relevant programmable liposome platforms with both the encapsulation stability of conventional formulations and the on-demand potency of free drug.