Photocatalytic protein labeling has become a powerful strategy for mapping proximity relationships inside cells, yet the field has remained heavily reliant on metal-based photoredox catalysts, reactive nitrenes, carbenes, and singlet oxygen. These tools work well in many contexts, but their chemical scope is narrow and their cellular compatibility is not always guaranteed. Expanding the reactive toolkit available to chemical biologists, particularly with fully organic systems that can operate deep inside living cells, remains a pressing challenge.
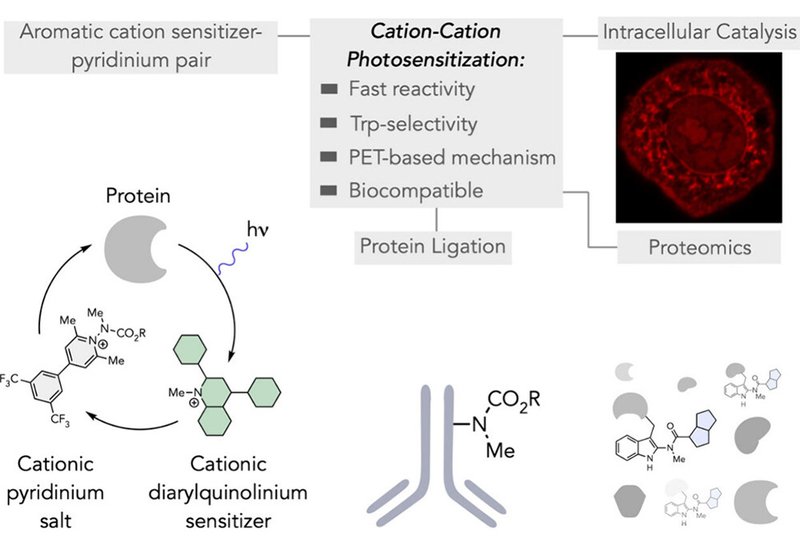
Researchers in the Taylor Group at the University of Arizona, published in J. Am. Chem. Soc., describe a new metal-free photosensitization strategy that pairs a 2,4-diaryl-N-methyl quinolinium sensitizer with an N-substituted pyridinium salt to achieve tryptophan-selective protein ligation driven by visible light. The approach extends an earlier platform from the same group for pyridinium-based tryptophan, Trp, ligation by introducing a catalytic sensitizer component, enabling the reactive pyridinium to be activated at low concentrations rather than requiring direct excitation of the stoichiometric reagent.
The team synthesized a small library of quinolinium sensitizers bearing varied aryl substituents and systematically screened them against model protein lysozyme under kinetically demanding conditions. Moderately electron-donating methoxy groups consistently outperformed both strongly donating dialkylamino groups and electron-withdrawing or heteroaromatic variants. The optimized sensitizer, compound 16 paired with pyridinium salt 20, achieved near-complete labeling of leuprorelin peptide at 10 mol% catalyst loading, and installed azide handles onto the therapeutic antibody trastuzumab with site-mapping pointing to W99 on the heavy-chain Fab region. Mechanistic perturbation studies established that photoinduced electron transfer drives the process: light-excited sensitizer 16 is reductively quenched by glutathione to generate a radical anion that undergoes single-electron transfer to pyridinium 20, producing a short-lived carbamoyl radical that reacts selectively with aromatic nucleophiles, overwhelmingly Trp.
Moving into complex biological systems, the researchers applied the 16/20 pair to HEK293T cell lysates and observed robust, dose-dependent labeling with measurable signal appearing within one to five minutes of irradiation. Chemical proteomics identified 319 enriched proteins and 615 modification sites, of which 521 resided on Trp, corresponding to 93% amino acid selectivity. Gene Ontology analysis highlighted significant enrichment of mRNA-binding proteins, and the team demonstrated that functionally unannotated Trp residues on proteins including Nup205, PPIF, and EIF4A1 could be captured. In live HEK293T cells, 101 proteins were enriched, with 57% annotated as nuclear by cellular component analysis, confirming that both reagents cross membranes and colocalize sufficiently to enable catalysis in nuclear compartments, a localization profile that contrasts with the mitochondrial preference typically assumed for hydrophobic aromatic cations.
The work establishes that cation-cation photosensitization is a viable and tunable design principle for intracellular chemical biology. Because the entire system consists of simple organic molecules requiring no metals, no genetic tags, and no specialized cellular engineering, it offers a straightforward route to Trp-centric proteomic profiling across structurally diverse biomolecules and cellular environments. The authors anticipate that the platform will find application in the catalytic synthesis of therapeutic protein conjugates and in mechanistic interrogation of biology in contexts where metal catalysts are impractical.