The RhoA G17V oncogenic mutant occupies a frustrating corner of the cancer target landscape. Found in nearly 70% of angioimmunoblastic T-cell lymphoma, AITL, cases, the variant abandons canonical nucleotide binding and instead latches onto the guanine nucleotide exchange factor Vav1, triggering phosphorylation of Tyr174 and hyperactivation of T-cell receptor signaling. The same structural features that make GTPases such precise biological switches, namely their rigid globular fold, shallow surface topology, and high-affinity nucleotide pocket, have blocked every conventional small-molecule campaign. Antibodies cannot cross the cell membrane to reach the intracellular target, leaving a compelling therapeutic rationale with no viable chemical matter to act on it.
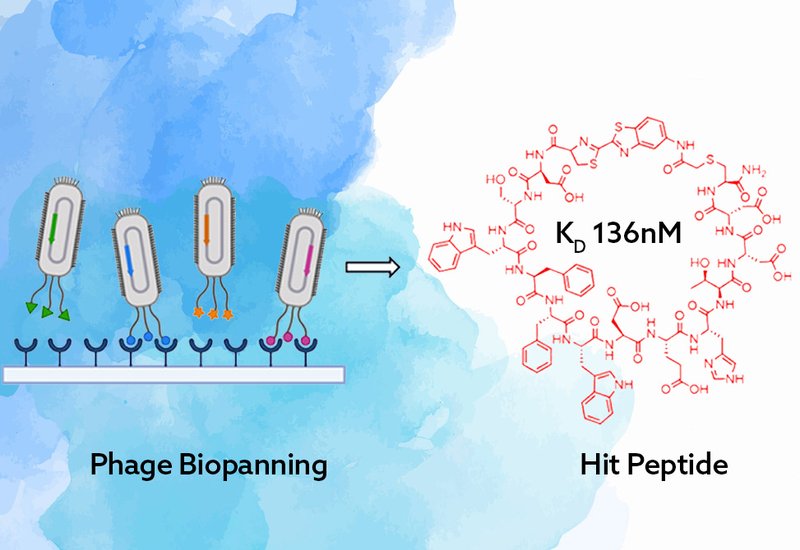
Researchers in the Liu-Hampton Lab at Texas A&M University, published in Biochemistry, addressed this gap by deploying two complementary macrocyclic phage display libraries against recombinant, biotinylated RhoA G17V. The first library used a genetically encoded acryloyl-lysine residue, AcrK, to cyclize a 10-mer peptide via a thioether bond between the AcrK side chain and an N-terminal cysteine. The second used a CAmCBT organic linker to cyclize a 12-mer library through cyanobenzothiazole condensation at the N-terminal cysteine and nucleophilic substitution at an internal cysteine. Both libraries underwent four rounds of high-stringency biopanning with alternating immobilization chemistries and progressively reduced target loading, followed by Illumina next-generation sequencing to map enrichment.
The 10-mer AcrK library returned moderate-affinity binders: RhoA2 at KD = 6.32 μM, RhoA3 at 27.33 μM, and RhoA5 at 10.69 μM as measured by biolayer interferometry. The 12-mer CAmCBT library performed at a different level. Peptide Z1, sequence CDSWFFWDEHTDDC, emerged as the top-ranked sequence across all three parallel biopanning strategies and bound RhoA G17V with a KD of 136 nM, the highest affinity reported for any macrocyclic peptide targeting this mutant. Selectivity profiling showed Z1 binds RhoA G17V with 4.7-fold higher affinity than wild-type RhoA, and 11.7-fold and 12-fold selectivity over KRAS and RAB7A, respectively. The linear counterpart of Z1 showed no detectable binding, confirming that macrocyclization is essential for the preorganized geometry driving target engagement.
To map the structural basis of binding, the team applied blind docking with GNINA 1.3 to an AlphaFold2-generated RhoA G17V model, clustered the resulting 100 conformations, and advanced eight representatives through 100 ns molecular dynamics runs, extending the most stable to 1000 ns. Conformation 1 settled into a site overlapping the GDP-binding pocket and the G17V mutation site, reaching an average binding free energy of −28.46 ± 16.05 kcal/mol. The MD trajectory revealed that residues F4, F5, and W6 of Z1 anchor a buried hydrophobic cluster, while E8 forms a directional hydrogen bond with RhoA D87 and D12 contacts K162. Simulations of Z1 docked to wild-type RhoA showed progressive dislocation after 800 ns and loss of most contacts, consistent with the lower experimental affinity and attributable to the absence of the Val17 side chain. Alanine scanning mutagenesis validated these predictions: single-residue substitutions W3A, F4A, F5A, W6A, E8A, and D12A each reduced affinity by more than 9-fold, while mutations at peripheral polar positions caused only 2- to 4-fold changes.
The convergence of phage selection, biolayer interferometry, long-timescale simulation, and residue-level mutagenesis establishes a coherent mechanistic picture and a reusable workflow. Z1 demonstrates that a well-chosen cyclization chemistry, combined with adequate library length and stringent alternating-resin biopanning, can break through the affinity ceiling that frustrated earlier efforts against mutant GTPases. The authors note that Z1 and related binders can serve as starting points for optimization toward cell permeability and intracellular delivery, and that the mutant-selective pharmacology opens a path to proximity-induced degradation strategies targeting RhoA G17V in AITL and related diseases driven by pathogenic GTPase mutations.