Xanthine oxidase catalyzes the final steps of purine catabolism, converting hypoxanthine and xanthine to uric acid. Dysregulation of this enzyme leads to excessive uric acid accumulation, driving hyperuricemia and gout pathogenesis. While synthetic inhibitors such as allopurinol and febuxostat effectively suppress xanthine oxidase activity, they engage in non-specific interactions with off-target proteins, potentially causing adverse effects including cardiovascular risks with long-term use. Peptide-based inhibitors offer an attractive alternative due to their high target specificity, excellent biocompatibility, and low toxicity. However, traditional discovery approaches relying on protein hydrolysis coupled with molecular docking remain constrained by limited structural diversity and poor correlation between predicted binding affinities and actual bioactivity.
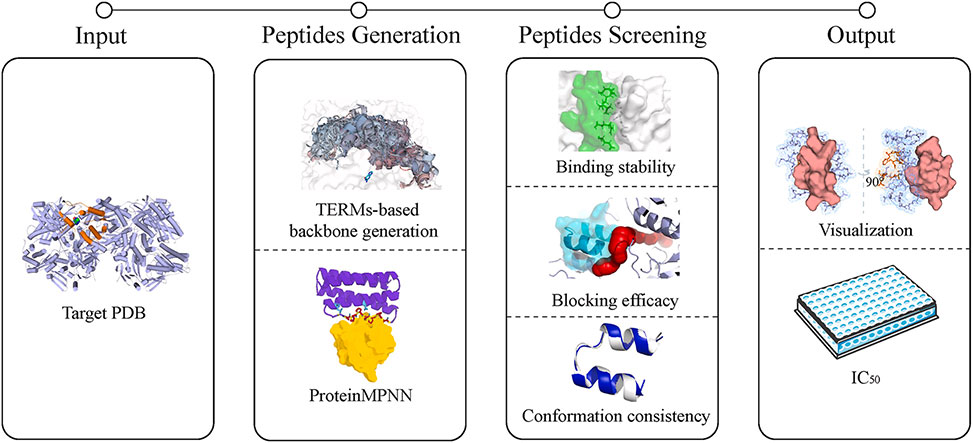
Researchers in the Guo Group at South-Central Minzu University, Wuhan, published in the European Journal of Medicinal Chemistry, developed a comprehensive computational framework for de novo design of xanthine oxidase inhibitory peptides. The pipeline combines tertiary motif-based backbone construction with the deep learning tool ProteinMPNN for sequence generation, followed by multi-tiered screening that integrates physics-informed methods with molecular dynamics simulations. A key methodological innovation replaces energy-minimized conformations with MD-stabilized conformations as the reference state for evaluating binding stability, blocking efficacy, and conformational consistency.
Abstract Figure
The backbone design process began by identifying 85 surface residues within 20 Å of the xanthine oxidase active site and generating structurally complementary peptide fragments as seeds, yielding 67,868 initial fragments. Fusion of these seeds produced 4000 candidate backbones, from which 88 met stringent selection criteria for potential contact number and proximity to the active site. Compared to 18 high-performance inhibitory peptides reported in recent literature, the designed backbones exhibited higher average potential contact numbers and shorter minimum distances to the catalytic center. The designed peptides averaged 16 amino acids in length with predominantly α-helical secondary structure at 70%, contrasting sharply with the positive controls that averaged only 6 residues and consisted primarily of random coils. ProteinMPNN then generated five sequences for each backbone, producing 440 candidates that underwent screening for solubility and toxicity using deep learning models. Energy minimization with Rosetta Relax evaluated binding free energy, while tunnel length calculations assessed the capacity to block substrate entry into the active site. AlphaFold3 predicted monomeric conformations to assess structural consistency between free and bound states, narrowing the pool to 10 candidates. Molecular dynamics simulations over 50 nanoseconds then evaluated these candidates under physiologically relevant conditions. Three peptides showing continuous RMSD increases and dissociation from the active pocket were eliminated. Among the remaining seven, peptide 1121_4 demonstrated the lowest conformational deviation between its monomeric state and MD-stabilized binding pose at only 0.9 Å RMSD. This 22-residue peptide adopts an αα-hairpin structure that forms a three-helix bundle arrangement with a surface helix of the xanthine oxidase active pocket, a topology known for conferring stability in protein-protein interactions.
Experimental validation confirmed the computational predictions. Peptide 1121_4 achieved an IC50 of 0.38 mM, representing a twofold improvement over FGGEH, the most potent previously reported xanthine oxidase inhibitory peptide with an IC50 of 0.81 mM under identical conditions. Surface plasmon resonance measurements revealed a dissociation constant of 5.17 μM, comparable to the clinically used inhibitor febuxostat at 2.7 μM. Cytotoxicity assays showed no observable toxicity across the tested concentration range. The framework, released as the DePocket pipeline on GitHub, establishes a scalable approach for enzyme-targeting peptide design. By demonstrating that MD-stabilized conformations provide superior screening accuracy compared to energy-minimized structures, this work offers a principled foundation for developing high-affinity peptide inhibitors across diverse enzymatic targets.
The Guo Group at South-Central Minzu University, Wuhan, China, focuses on fundamental theory and applied technology in microbial resource engineering, systematically exploring functional discovery and targeted regulation of probiotics, prebiotics, and bioactive peptides, integrating a multi‑level platform covering computational biology, genetic engineering, cellular models, and animal studies. Their research spans rational design of engineered probiotics, synergy analysis of probiotics/prebiotics/postbiotics via gut microbiota–host interactions, intestinal homeostasis models, organoids, metabolic phenotyping, and AI‑driven discovery of active proteins/oligopeptides using machine learning and target prediction. The work highlighted here presents a development of a comprehensive computational framework for the de novo design of xanthine oxidase inhibitory peptides, enabling rational discovery of novel bioactive sequences with targeted enzymatic inhibition.
