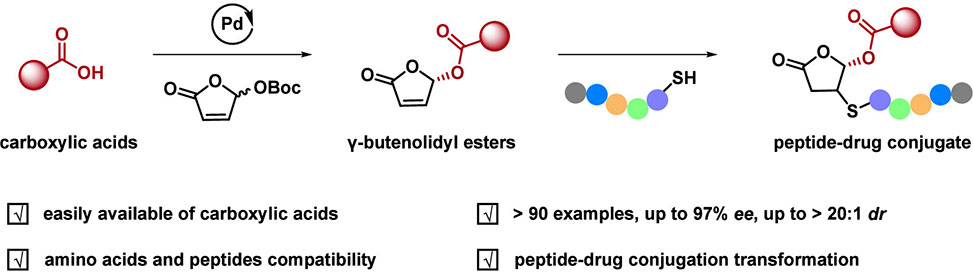
Carboxylic acid groups frequently serve as essential pharmacophores in drug molecules, yet their high polarity limits membrane permeability and can reduce bioavailability. Prodrug strategies address this challenge by masking the carboxylic acid as an ester that improves lipophilicity for cellular uptake, then releases the active compound through enzymatic hydrolysis or spontaneous conversion. Many ester-based prodrugs have reached clinical use, including Enalapril, Pivampicillin, and Azilsartan Medoxomil. As prodrug structures grow more complex, stereochemistry has emerged as a critical design parameter. Enantiomers of chiral ester prodrugs often exhibit distinct metabolic profiles and biological activities, interacting differently with esterases to produce variations in release kinetics. However, existing methods for synthesizing chiral prodrug scaffolds typically require prior activation of carboxylic acids into acyl chlorides or anhydrides, adding synthetic steps that limit practical application.
A team of researchers at the Beijing University of Chemical Technology, supervised by Professor Duanyang Kong, published in Organic Letters, developed a palladium-catalyzed asymmetric allylic alkylation that directly converts carboxylic acids into chiral γ-substituted butenolide esters without preactivation. The reaction couples γ-OBoc-butenolides with carboxylic acids using a catalyst generated in situ from Pd2(dba)3 and a Trost ligand. Optimization established that dichloromethane at room temperature with 2 mol% catalyst loading delivered optimal results, achieving 91% yield with 93% enantiomeric excess for the model substrate phenylacetic acid. The addition of potassium carbonate proved beneficial for electron-deficient substrates by generating more nucleophilic carboxylate anions that facilitate the key bond-forming step.
Abstract Figure
The substrate scope proved remarkably broad. Arylacetic acids bearing electron-withdrawing groups including fluoro, chloro, bromo, nitro, cyano, and sulfonyl substituents all delivered products with excellent enantioselectivity ranging from 90–97% ee. Electron-rich substrates with methyl, methoxy, and protected amino groups performed equally well. Potentially reactive functionalities such as boronic ester and terminal alkyne remained intact under the mild conditions. Heteroaromatic carboxylic acids derived from thiophene, furan, benzofuran, and indole scaffolds underwent smooth transformation with 91–94% ee. The reaction accommodated secondary and tertiary carboxylic acids alongside primary substrates. Pharmaceutically relevant acids including Diclofenac, Naproxen, Flurbiprofen, Ketoprofen, Ibuprofen, and Febuxostat all converted efficiently to their corresponding chiral butenolidyl esters. Benzoic acids, alkyl acids, and polyunsaturated substrates such as sorbic acid and retinoic acid likewise proved compatible. For amino acid substrates, the method delivered products with good yields and diastereoselectivities from 7:1 to greater than 20:1 dr. Proteinogenic amino acids spanning phenylalanine, leucine, valine, methionine, cysteine, proline, tryptophan, and lysine all participated successfully, as did nonproteinogenic variants and short oligopeptides.
The chiral butenolidyl products serve dual purposes as prodrug scaffolds and conjugation handles. The α,β-unsaturated lactone functions as a Michael acceptor for cysteine thiols under mild, biocompatible conditions. The researchers demonstrated peptide-drug conjugate synthesis by reacting chiral butenolidyl esters with cysteine-containing peptides in aqueous buffer at room temperature, generating ten distinct conjugates. This reactivity positions the butenolidyl scaffold as a novel alternative to maleimides for constructing peptide-drug and antibody-drug conjugates. Mechanistic studies revealed that the transformation proceeds through kinetic resolution, with palladium generating a π-allyl intermediate that undergoes out-of-plane nucleophilic attack by the carboxylic acid. The Trost ligand creates a chiral environment where one transition state experiences favorable steric interactions while the competing pathway encounters repulsion from the ligand's aromatic rings, accounting for the observed enantioselectivity. This asymmetric platform expands the molecular toolbox for precision medicinal chemistry by enabling direct, stereocontrolled access to chiral prodrugs and bioconjugates from unactivated carboxylic acids.
