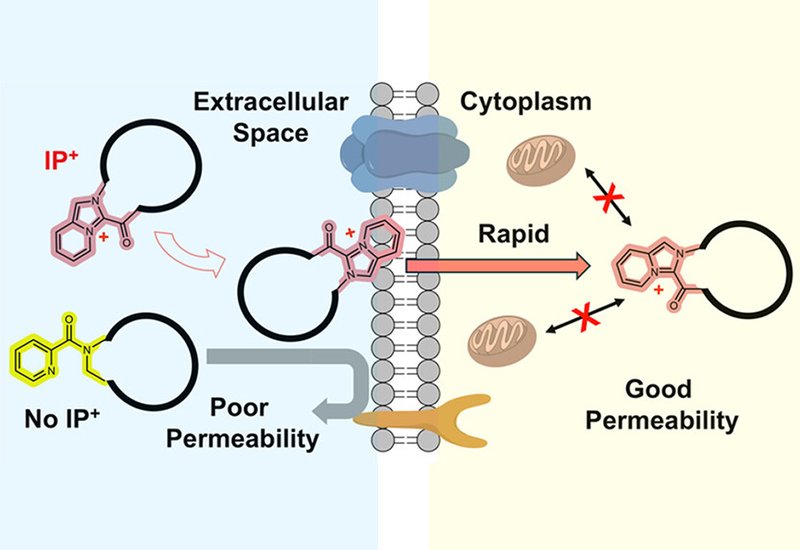
Macrocyclic peptides have emerged as promising therapeutic candidates because they bind difficult protein targets with high affinity and selectivity. Yet their application to intracellular targets remains limited by poor passive membrane permeability, which stems from the well-hydrated amide N–H groups that resist diffusion through lipid bilayers. Most efforts to overcome this barrier have focused on emulating natural products such as Cyclosporine A, which relies on extensive N-methylation, intramolecular hydrogen bonds, and highly hydrophobic side chains to achieve cell permeability. While effective, this strategy narrows chemical space and structural diversity available for drug discovery.
Researchers in the Kodadek Group at The Herbert Wertheim UF Scripps Institute for Biomedical Innovation and Technology, published in the Journal of the American Chemical Society, report a detailed cellular analysis of macrocyclic peptides containing an imidazopyridinium, IP+, unit. The permanent positive charge of this moiety was hypothesized to attract the peptide to the phospholipid bilayer surface, after which the hydrophobic nature of the ring system might facilitate movement through the membrane. The team previously demonstrated that many IP+ macrocyclic peptides with molecular weights ranging from 600 to 1200 Da traversed an artificial membrane at rates typical of much smaller, drug-like molecules in the parallel artificial membrane permeability assay. The current study evaluates whether this behavior translates to living mammalian cells.
The chloroalkane penetration assay, CAPA, was used to measure relative cell permeability. Cells expressing HaloTag protein were exposed to macrocyclic peptide–chloroalkane conjugates for defined periods. Unoccupied HaloTag was then labeled with a fluorescent chloroalkane probe, and residual fluorescence was quantified. Lower fluorescence indicates greater cytosolic exposure of the test molecule. A five-residue IP+ macrocyclic peptide, MP1, containing tryptophan and phenylalanine, exhibited a CP50 of 0.76 μM, only four-fold higher than the drug-like small molecule JQ-1. A control peptide with a picolinic amide linkage in place of the IP+ ring, MP2, was more than 40-fold less permeable. Placement of the IP+ unit on a side chain rather than in the macrocyclic backbone, MP3, yielded intermediate permeability. A dual IP+ peptide, MP4, incorporating the charged ring at both the ring junction and on a side chain, displayed a CP50 of 0.25 μM, nearly equivalent to JQ-1 and more than 120-fold better than MP2. Across a series of diverse macrocyclic peptides, including those containing arginine, 3-pyridylalanine, and carboxylic acid residues, IP+ incorporation consistently improved permeability. Even larger peptides, with molecular weights exceeding 1300 Da and multiple polar side chains, benefited from IP+ grafting, with four- to six-fold improvements in CP50 values. Elaboration of the IP+ ring with additional substituents modestly reduced permeability relative to the unsubstituted heterocycle, suggesting the parent scaffold is optimal.
Live-cell imaging with MP1-bodipy revealed immediate, uniform staining of the plasma membrane within one minute of exposure, followed by rapid cytosolic accumulation over the next 40 minutes. Time-dependent CAPA experiments showed that MP1–chloroalkane alkylated half of the cytosolic HaloTag protein within 30 minutes and nearly all of it within two hours, a rate approximately seven- to ten-fold slower than JQ-1 but far faster than typical cell-penetrating peptides, which rely on endocytosis and often require hours. Treatment with sodium azide and 2-deoxyglucose, classical inhibitors of energy-dependent endocytosis, had no effect on MP1 entry at multiple concentrations, consistent with passive diffusion. Colocalization studies using MitoTracker Deep Red confirmed that MP1-bodipy does not accumulate at mitochondrial membranes, unlike many positively charged molecules. At later time points, bubble-like structures became apparent in living cells; these collapsed into fluorescent puncta upon fixation, a known artifact for small, mobile molecules. Cotreatment with CPP12, a well-characterized cell-penetrating peptide that enters via vesicle budding and collapse, revealed distinct kinetics and minimal early colocalization, further supporting passive membrane diffusion as the primary mechanism for IP+ macrocyclic peptides.
To test whether IP+ incorporation could rescue the cellular activity of an existing protein-binding macrocyclic peptide with poor permeability, the team modified UNP-6457, a nine-residue antagonist of the p53–MDM2 interaction with picomolar in vitro potency but no cellular activity. Analysis of the cocrystal structure revealed that the tryptophan residue is solvent-exposed and does not contact MDM2. Replacing this residue with an IP+ unit generated MP23, which retained strong MDM2 binding in vitro with only modest loss of affinity. MP23 displayed measurable cytotoxicity to MCF-7 breast cancer cells, with an IC50 of approximately 41 μM, whereas UNP-6457 showed no effect. CAPA confirmed that MP23 was ten-fold more permeable than UNP-6457. An alternative design, MP24, in which the IP+ unit replaced the triazole at the ring closure, exhibited improved permeability but minimal cytotoxicity, consistent with its reduced affinity for MDM2, likely due to altered macrocycle conformation. The strong correlation between MDM2 binding and cytotoxicity supports on-target activity for MP23. None of the IP+ macrocyclic peptides tested, even at 100 μM, were toxic to HEK293 cells.
These results establish that IP+ incorporation is an effective and generalizable strategy for improving the passive cell permeability of macrocyclic peptides. The approach tolerates polar and anionic side chains, works with peptides ranging from 600 to over 1300 Da, and functions whether the IP+ unit is positioned in the macrocyclic backbone or on a side chain. Entry into cells occurs rapidly, via an energy-independent mechanism consistent with passive diffusion, without mitochondrial accumulation or cellular toxicity. The transformation of a membrane-impermeable MDM2 antagonist into a bioactive inhibitor demonstrates the practical utility of this design principle for developing probe molecules and drug candidates targeting intracellular proteins.