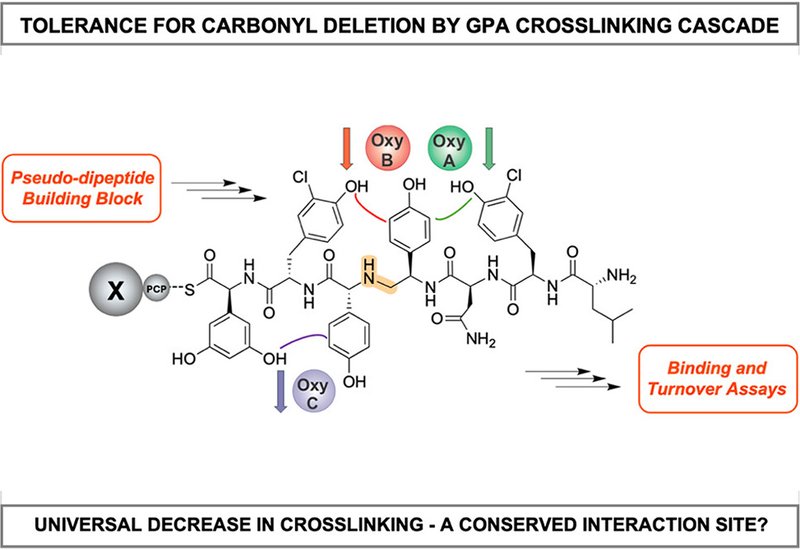
Glycopeptide antibiotics, GPAs, including vancomycin and teicoplanin represent critical last-resort treatments against multidrug-resistant bacterial infections. These complex natural products derive their antimicrobial potency from a rigid, highly cross-linked peptide core that enables precise binding to bacterial cell wall precursors. The cross-links are installed by a carefully choreographed cascade of cytochrome P450 enzymes, collectively called Oxy enzymes, which operate while the peptide remains tethered to its biosynthetic machinery. Synthetic chemists have demonstrated that modifying the peptide backbone with methylene linkages can restore activity against resistant bacterial strains, yet whether biosynthetic enzymes could tolerate such modifications remained unexplored. Understanding this tolerance could unlock biocatalytic routes to modified GPAs, circumventing the formidable synthetic challenges of total chemical synthesis.
Researchers supervised by Professors Julien Tailhades and Max J. Cryle from Monash University, Australia, published in ACS Chemical Biology, have developed a chemoenzymatic strategy to test whether Oxy enzymes could cross-link peptides containing methylene backbone modifications. They synthesized a solid-phase peptide synthesis compatible methylene pseudodipeptide that replaced the central D-hydroxyphenylglycine-D-hydroxyphenylglycine, D-Hpg-D-Hpg, building block in GPA peptides. This dipeptide mimetic was incorporated into peptide substrates resembling different GPA types, including vancomycin-like, teicoplanin-like, and avoparcin-like sequences. The modified peptides were loaded onto peptidyl carrier protein, PCP, domains and subjected to the sequential action of four different Oxy enzymes: OxyB, which installs the initial cross-link; OxyE for certain GPA types; OxyA, which forms the second cross-link; and OxyC, which completes the tricyclic core structure. Each enzyme's activity was monitored by mass spectrometry analysis of cross-linked products.
The methylene backbone modification devastated enzymatic activity across the entire Oxy cascade. Control peptides containing normal amide bonds showed robust cross-linking, with the chlorinated vancomycin-type substrate achieving greater than 50% conversion to the fully tricyclic product. In stark contrast, methylene-modified substrates suffered dramatic reductions in cross-linking efficiency: OxyB activity dropped to 20%, OxyA to 12.7%, and OxyC to 20.5% compared to controls. This created a compounding effect where diminishing substrate availability at each step led to vanishingly small amounts of final cross-linked products. Notably, the effect was general rather than enzyme-specific, unlike previous modifications that selectively impaired individual Oxy enzymes. Binding studies using ultraviolet-visible spectroscopy revealed that methylene substrates maintained similar binding affinity to OxyB but showed dramatically reduced binding efficiency, as measured by their ability to trigger the characteristic low-spin to high-spin heme transition that accompanies substrate binding. NADH consumption experiments confirmed that reduced cross-linking activity correlated with diminished enzyme turnover rates, while hydrogen peroxide formation remained unchanged, ruling out uncoupling as the primary cause of reduced activity.
These findings reveal that GPA biosynthetic enzymes are exquisitely sensitive to backbone carbonyl deletion, suggesting these groups play essential roles in substrate positioning within enzyme active sites. The universal impairment across all Oxy enzymes indicates that backbone carbonyls likely participate in critical hydrogen-bonding networks that facilitate proper substrate binding and catalytic turnover. This discovery limits the potential for using biosynthetic approaches to produce methylene-modified GPAs, despite their promising activity against resistant bacteria. The work underscores how precisely tuned natural biosynthetic machinery can be and highlights the need for detailed structural characterization of enzyme-substrate complexes to guide future engineering efforts. For the development of next-generation antibiotics, these results suggest that achieving backbone modifications may require continued reliance on synthetic chemistry rather than biocatalytic methods, at least until the structural basis for substrate recognition is better understood and enzyme engineering strategies can be developed.