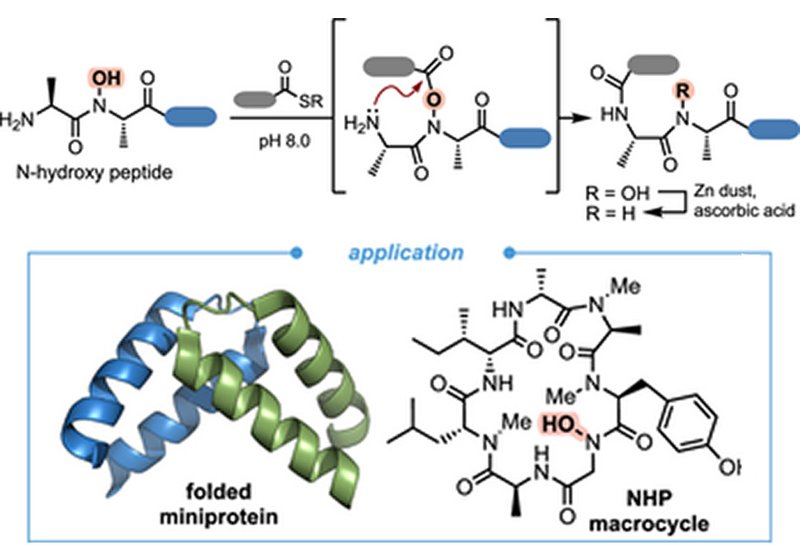
Chemical ligation of unprotected peptide fragments in aqueous media has long depended on native chemical ligation, NCL, which exploits a thiol at the N-terminus of the nucleophilic fragment to drive transthioesterification and S→N acyl transfer at cysteine residues. That constraint is limiting: nearly one in five residues N-terminal to Cys are β-branched, and the synthesis of thiolated building blocks for alternative junctions can require eight or more synthetic steps. The field has made steady progress through auxiliary-based strategies and side-chain hydroxamate capture, yet achieving efficient amide-bond formation at truly arbitrary junctions remains an open challenge.
Ph.D. student Natalia Cano-Sampaio and Professor Juan R. Del Valle at the University of Notre Dame, published in Chemistry – A European Journal, describe a general ligation strategy built on the unique reactivity of backbone N-hydroxy peptide, NHP, fragments. Rather than relying on side-chain identity, the approach positions a hydroxamate within the peptide backbone at the penultimate residue of the nucleophilic fragment, enabling O-acyl capture and O→N acyl transfer in aqueous buffer. Protected N-hydroxy dipeptide building blocks were prepared via Fukuyama N-hydroxylation and condensation with Fmoc amino acid chlorides; on-resin submonomer assembly provided an alternative route for hGly-containing fragments. Global deprotection and RP-HPLC purification delivered eleven NHP substrates ready for ligation screening.
Systematic evaluation of residue scope revealed broad tolerance at every position tested. Charged, β-branched, aromatic, and N-methylated residues at the nucleophilic N-terminus all participated in ligation with peptide thioesters at pH 8.0, with ligation rate increasing at higher pH consistent with hydroxamate deprotonation accelerating O-acylation. A control peptide lacking the N-hydroxyl group produced only trace product, confirming the mechanistic centrality of the hydroxamate. Preparative-scale ligations across ten distinct junctions gave isolated yields ranging from 23% to 80%, including at junctions that would otherwise require multi-step syntheses of thiolated analogs of Lys, Arg, and Phe. Subsequent reduction with zinc powder in aqueous ascorbic acid cleaved the N–O bond and restored the native amide, furnishing eupeptidic products in 48–71% isolated yield.
The method's scope was demonstrated across three demanding targets. An hGly-containing collagen mimetic peptide assembled by NHP ligation showed a melting temperature of 38.8°C, nearly 6°C higher than the parent Gly-containing variant at 33.1°C, revealing that a single backbone N-hydroxylation at the Yaa position significantly stabilizes the triple-helical collagen fold. For the Sam68 homodimerization domain, a 37-residue miniprotein, introduction of hAla at position 121 abolished folding entirely, whereas zinc-mediated reduction to the native amide restored the α-helical CD signature and a Tm of 42.4°C matching the recombinant protein. In macrocycle synthesis, an intramolecular NHP ligation between N-methylated terminal residues completed talarolide A in 27% overall yield as a single atropisomer, matching the 1H NMR of the natural product in every respect and avoiding the 2:1 atropisomeric mixture produced by prior HATU-mediated cyclization.
Together, these results position backbone NHP ligation as a versatile and sequence-agnostic complement to thiol-based ligation chemistry. Because the hydroxamate is embedded in the backbone rather than derived from any particular side-chain, the strategy sidesteps the residue-specific constraints of NCL and its thiolated-building-block extensions. The ability to toggle conformational stability through N-hydroxylation and then restore native structure by reductive N–O cleavage adds a controllable dimension to miniprotein and macrocycle design that earlier ligation platforms do not offer. Extension to the circularization of larger proteins and to the synthesis of other NHP natural products now appears within reach.