Hypertension remains the leading cause of premature death worldwide, yet one in five affected adults achieve adequate blood pressure control. Angiotensin-converting enzyme inhibitors represent one of the most prescribed antihypertensive drug classes, targeting the renin-angiotensin system that regulates vascular tone. When blood pressure drops, the kidney releases renin to convert angiotensinogen into angiotensin I, which ACE then processes into angiotensin II, a potent vasoconstrictor. ACE also degrades vasodilator peptides including bradykinin, making it a dual-action target for blood pressure management. However, existing ACE inhibitors cause dry cough in many patients due to bradykinin accumulation, frequently leading to treatment abandonment. This adverse effect arises because current drugs lack selectivity between the enzyme's two catalytic domains. The C-domain primarily generates angiotensin II when compared to the N-domain, while both domains degrade bradykinin, suggesting that C-domain selective inhibitors could reduce side effects while maintaining antihypertensive efficacy.
Researchers at Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil, under the leadership of Professor Walter Orlando Beys-da-Silva, published in ACS Medicinal Chemistry Letters, identified two novel ACE-inhibitory peptides through virtual screening of an in-house peptide database. Traditional discovery of bioactive peptides from natural sources such as marine organisms, plant extracts, or animal tissues requires substantial experimental effort for preparation, characterization, and isolation. Molecular docking offers an alternative approach capable of discriminating active compounds from inactive decoys when correctly implemented, thereby accelerating the identification of promising candidates for experimental validation. The team applied this computational strategy to screen their peptide library against the crystal structure of testicular ACE, which corresponds to the C-domain sequence.
Abstract Figure
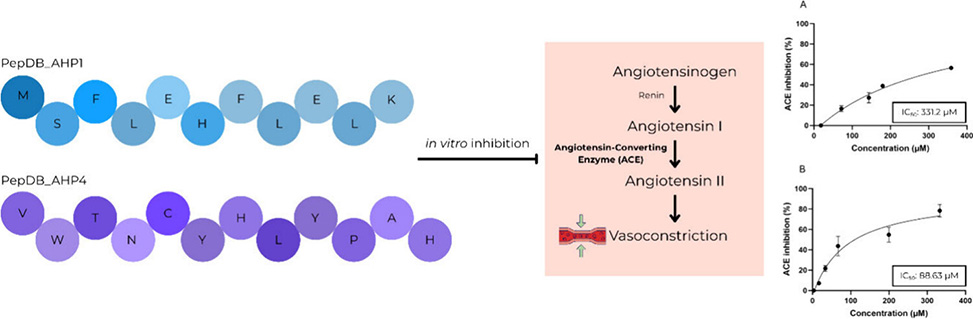
Two peptides emerged from the virtual screen: PepDB_AHP1 with sequence MSFLEHFLELK and PepDB_AHP4 with sequence VWTNCYHLYPAH. Molecular docking revealed that both peptides interact with residues at the enzyme's active site including His353, Ala354, and Val380. PepDB_AHP1 achieved a docking score of −9.4 kcal/mol through interactions spanning all three binding pockets: hydrogen bonds with Tyr523 and Ala354 at the S1 pocket, hydrogen bonding with Glu162 at the S1′ pocket, and π-stacking with His353 at the S2′ pocket. The peptide also forms salt bridges with His387, which coordinates the catalytic zinc ion. PepDB_AHP4 scored −9.1 kcal/mol and established hydrogen bonds with Tyr523, Ala354, His353, and Glu411, the latter also coordinating zinc. Both peptides contact Val379 and Val380 through hydrophobic interactions, residues that are exclusive to the C-domain and support the hypothesis of domain selectivity.
Enzymatic inhibition assays confirmed the computational predictions. PepDB_AHP4 demonstrated an IC50 of 88.63 μM while PepDB_AHP1 yielded 331.2 μM. The nearly four-fold difference in potency correlates with hydrogen bonding patterns: PepDB_AHP4 forms 12 hydrogen bonds among its 16 total interactions, whereas PepDB_AHP1 establishes only 5 hydrogen bonds out of 22 interactions. Literature precedent supports hydrogen bonding as a critical determinant of ACE inhibitor potency. The C-terminal residue also influences activity, as positively charged amino acids enhance binding while dicarboxylic residues prove less favorable. PepDB_AHP1 terminates with lysine but contains a penultimate glutamate that may diminish potency, while PepDB_AHP4 ends with histidine. Both peptides display hydrophobic character through multiple aromatic and aliphatic residues, consistent with structural requirements for ACE engagement. Although the IC50 values fall in the micromolar range rather than the nanomolar potency of optimized therapeutics, these peptides establish validated starting points for medicinal chemistry optimization. Strategies including sequence truncation, incorporation of noncanonical amino acids, addition of functional groups, and cyclization could enhance both potency and proteolytic stability, advancing these scaffolds toward C-domain selective antihypertensive agents with improved side effect profiles.



